| Prev | ICM User's Guide 9.7 Regularization | Next |
| Available in the following product(s): ICM-Pro |
Regul stands for Regularization which is a procedure for fitting a protein model with the ideal covalent geometry of residues (as represented in the icm.res residue library) to the atom positions of a target PDB structure.
Regularization is a procedure for fitting a protein model with the ideal covalent geometry of residues (as represented in the icm.res residue library) to the atom positions of a target PDB structure (usually provided by X-ray crystallography or NMR). Regularization is required because the experimentally determined PDB-structures often lack hydrogen atoms and positional errors may result in the unrealistic van der Waals energy even if these structures were energetically refined (since the refinement of the crystallographic structures typically ignores hydrogen atoms and employs different force fields). The following steps are required to create the regularized and energy refined ICM-model of an experimental structure.
To use the regularization protocol:
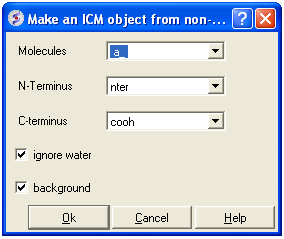
- MolMechanics/Regularization and a data entry box as shown below will be displayed.
- Enter the molecule you wish to refine.
- Choose which kind of N and C- terminus
- Choose whether you wish to include water molecules.
- Choose to run in background if the structure is very large.
Once the refinement is complete a new ICM object will be displayed in the ICM workspace called object_name_reg
| Prev Find PDB Loop Segment | Home Up | Next Refine SC |