| Prev | ICM User's Guide 3.7 Preferences | Next |
[ Bonds Preferences | Directories Preferences | Graphics Preferences | GUI Preferences | GUI Prefs | Image Preferences | Font Pref | Plot Preferences | Ribbon Preferences | Shell Preferences | System Preferences ]
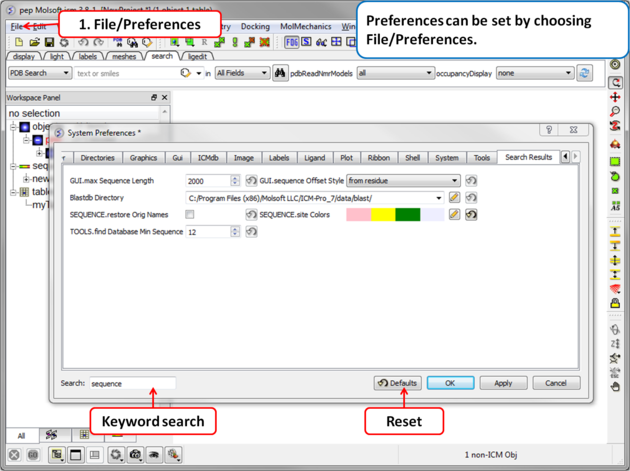
Your ICM preferences can be changed by:
- Select File/Preferences.
| NOTE: There is a "Reset to Default" button in case you make any changes you are not happy with and also a search option. |
3.7.1 Bonds Preferences |
To change Bond Preferences:
- Select File/Preferences.
- Choose the Bonds tab.
GRAPHICS.ballStickRatio - A default ratio of ball and stick radii. This ratio is applied when the styles are switched from the GUI xstick toolbar. Default (1.4)
GRAPHICS.hbond Ball Period - Default (3)
GRAPHICS.hbondMinStrength - parameter determines the hbond strength threshold for hbond display. The strength value is between 0. and 2. By changing 1. to 0.2 you will see more weak hydrogen bonds. Default: (1).
GRAPHICS.hbondStyle - determines the style in which hydrogen bonds are displayed. Here hbond-Donor, Hydrogen, and hbond-Acceptor atoms will be referred to as D, H and A, respectively,
GRAPHICS.hetatmZoom - The default ball and stick radii of a ligand can be different by the GRAPHICS.hetatmZoom factor. This makes a better ligand view since the ligand stands out from the surrounding protein atoms.
GRAPHICS.stickRadius - radius (in Angstroms) of a cylinder displayed as a part of stick or xstick graphical representation of a molecule. Individual (residue-wide) control of stick radii.
GRAPHICS.xstick Backbone Ratio - Default (1.2)
GRAPHICS.xstick Style - xstick style
wireBondSeparation the distance between two parallel lines representing a chemical double bond if wireStyle = "chemistry". Default (0.2 Angstroms).
GRAPHICS.distance Label Drag - enable distance label dragging
GRAPHICS.hbondAngleSharpness determines how the strength depends on the D-H...A(lone pair) angle. The preference can be found the general Preferences menu Default (1.7)
GRAPHICS.hbond Ball Style even, by atom size, by energy or telescopic
GRAPHICS.hbond Rebuild
GRAPHICS.hbondWidth relative width of a displayed hbond .
GRAPHICS.hydrogenDisplay determines the default hydrogen display mode for the display command.
GRAPHICS.hydrogenDisplay = "polar" 1 = "all" # all hydrogens are shown 2 = "polar" <-- current choice # polar displayed, the non-polar hidden 3 = "none" # no hydrogens are displayed
GRAPHICS.wire Width - relative width of wire Default (1)
GRAPHICS.xstick Hydrogen Ratio - Default (0.5)
GRAPHICS.xstick Vw Ratio - Default (0.6)
Wire Style - change the default wire style
GRAPHICS.hbondStyle = "dash" 1 = "wire" # Just a line 2 = "chemistry" # shows different types of chemical bonds. 3 = "tree" # shows a directed graph of the ICM-molecular tree 4 = "aromatic" #
3.7.2 Directories Preferences |
DIRECTORIES TAB:
To change Directory Preferences:
- Select File/Preferences.
- Choose the Directories tab.
Within this tab you can select the default directories for:
FILTER.gz, FILTER.uue, FILTER.Z, Filter.zip allows you to read compressed files .gz, .uue, .Z, and, .zip files automatically leaving the compressed file intact.
PDB Directory Style - The style of your Protein Data Bank directory/directories. ICM will understand all of the listed styles, including distributions with compressed *.gz , *.bz2 and *.Z fil es
BlastDB Directory - return directory with Blast-formatted sequence files for ICM sequence searches.You can download Blast formatted databases from here ftp://ftp.ncbi.nih.gov/blast/db/
Dock Directory - Default directory for storing docking files.
CCP4 Directory
Editor - Select a default text editor
Inx Directory - location of stored index (*.inx) files.
Log Directory - when you quit an icm-session, a _seslog.icm file is automatically stored. If the s_logDir variable is empty, it is stored to the s_userDir + "/log/" directory. However one can redirect it to the current working directory ( "." ) or any other directory.
Output Directory -
PDB Directory - directory containing the PDB database of 3D structures. These files can also be easily downloaded directly from the PDB site if the variables are set as in the example below. PDB distributions can exist in several styles (all files in the same directory, or divided etc.).
PDB Directory FTP
PDB Directory Web
Projects Directory - Select the default location for storing ICM projects. Save your data in an ICM project. It is a convenient way of keeping all your structures, alignments, tables, docking results etc... in one place. A description on how to save an ICM project is described in the GUI Basics section of this manual.
Prosite Dat - location of the prosite.dat file a dictionary of protein sites and patterns, (Copyright by Amos Bairoch, Medical Biochemistry Department, University of Geneva, Switzerland).
Ps Viewer - Select a postscript viewer
Swissprot Dat - location of swissprot.dat file
Temp Directory - scratch directory for temporary files ( some montecarlo files will be saved there ).
Uniprot Dat - location of uniprot.dat file
XPDB Directory - Path to the ICM XPDB database of compact binary ICM objects which are annotated with the site information. The advantage of the XPDB database is the speed of reading and smaller size than PDB. XPDB entries are read about 80 times faster!
TOOLS.default ChemDB
TOOLS.eds Directory
TOOLS.pdb Read Nmr Models
1. = "first" : reads only one model from a multi-model (e.g. NMR) pdb file 2. = "all" : reads all models from a multi-model (e.g. NMR) pdb file and creates a separate object for each of them 3. = "all stack" : creates one object and loads all other models as a stored cartesian stack
3.7.3 Graphics Preferences |
To change Graphics Preferences:
- Select File/Preferences.
- Choose the Graphics tab.
Atom Single Style - display style of isolated atoms in the wire mode.
1. "tetrahedron" 2. "cross" 3. "dot"
GRAPHICS.clash Style - choose clash length, strain or length.
GRAPHICS.clip Grobs - enable grob clipping.
GRAPHICS.clip Static -
GRAPHICS.grobLineWidth - relative width of displayed lines of 3D meshes ( grobs ). Also affects the interatomic distance display.
GRAPHICS.lightPosition - X ,Y and Z posiion of the light source in the graphics window. The X and Y coordinates are usually slightly@@ beyond the [-1. 1] range where [-1.,1.] is the size of the window, and the Z position is perpendicular to the screen and is set to 2. (do not make it negative).
GRAPHICS.occupancyDisplay preference controlling if and how the partical or zero atom occupancies are displayed. The abnormal occupanices are shown as circles around atoms. These following values are allowed.
1. = "none" # nothing is displayed 2. = "circle" # a circle is displayed 3. = "label" # a circle and a lable with the value (zero values are not shown)
GRAPHICS.quality - integer parameter controlling quality (density of graphical elements) of such representations as cpk, ball, stick, ribbon . Do not make it larger than about 20 or smaller than 1.
GRAPHICS.ruler Style - change ruler from center to side
GRAPHICS.stereoMode - 1. "up-and-down", 2. "line interleaved" 3. "in-a-window"
- a simple hardware stereo mode for workstations with a horizontal frame splitter.
- In the "up-and-down" mode a longer frame with two stereo images on top of each other is generated and the two halves are then superimposed with the splitter. This mode does not require anything from a graphics card, but does require a frame splitter. A frame splitter box was connected between a monitor and a graphics card output. This mode has an unpleasant side effect, the rest of the screen (beyond the OpenGl window) becomes stretched and the lower part of the screen is superimposed on the top half.
- The "line interleaved" mode can be used with a new type of frame splitter at the line level. In this case the odd lines from one stereo-image are interleaved with the even lines of another. The side-effect of this mode is that the intensity is reduced in half since at each moment one sees only one half of the lines. The splitter device for this mode can be purchased from Virex (www.virex.com). This mode produces a dark stereo image but is easily available (requires stereo goggles, e.g. from Virex).
- The "in-a-window" mode is used in SGI workstations and in a Linux workstation with an advanced graphics card supporting a quad graphics buffer. In this mode the hardware stereo regime applies only to an OpenGl window. This is the best mode but it requires an expensive graphics card (plus the stereo goggles).
GRAPHICS.surfaceDotDensity - Determines the number of dots per square Angstrom on the graphical solvent accessible surface.
GRAPHICS.surfaceProbeRadius - An increment to the van der Waals radii of atoms at thich the dotted atomic surface is calculated. It is used by the display surface command to display dotted van der Waals surface. If the GRAPHICS.surfaceProbeRadius is set to 1.4 the surface becames equivalent to the solvent accessible surface with a probe of 1.4A
GROB.arrowRadius - a real arrow radius in Angstoms used by the Grob( "ARROW", R_ ) function. Default: 0.5.
GROB.contourSigmaIncrement - a real increment in the sigma level used to re-contour an electron density map using the make grob m_eds add r_increment command. This parameter is used in the GUI when plus and minus are pressed.
GROB.relArrow Size - a real ratio of the arrow head radius to the arrow radius. This parameter is used by the Grob( "ARROW", R_ ) function. Default: 3.0.
shineStyle - defines how solid surfaces of cpk , skin and grobs reflect light. Possibilities:
1. "white" <- default 2. "color"
The first option gives a more shiny and greasy look.
GRAPHICS.center Follows Clipping - determine the function of center button.
GRAPHICS.clashWidth - relative width of a displayed clash .
GRAPHICS.clip Skin - enable skin clipping.
GRAPHICS.displayMapBox - controls if the bounding box of a map is displayed
GRAPHICS.light - a rarray of 13 elements between 0. and 1. which controls the main properties of lighting model in GL.
GRAPHICS.mapLineWidth - relative width of lines and dots of a displayed map.
GRAPHICS.occupancy Radius Ratio - preference controlling the radius of the partical or zero atom occupancies
GRAPHICS.resize Keep Scale
GRAPHICS.selectionStyle - preference for the style in which the graphical selection is shown. The preference may have the following values.
GRAPHIC.store Display - maintains representation and coloring for an object.
GRAPHICS.surfaceDotSize - Determines the size of the dot on the solvent accessible graphical surface.
GRAPHICS.transparency - Two parameters regulating the transparency of grobs.
GROB.atomSphereRadius - default radius (in Angstroms) which is used to select a patch on the surface of a grob.
GROB.relArrowHead - a real ratio of the arrow head radius to the arrow radius.
lineWidth - the real width of lines used to display the wire representation of chemical bonds.
3.7.4 GUI Preferences |
GUI TAB:
The options contained within the Preferences/Gui tab are described below.
GRAPHICS.alignment Rainbow - This option controls how alignments are colored by default.
GRAPHICS.NtoC Rainbow - Controls the coloring of structural representation from the N-terminal to the C-terminal
GRAPHICS.rocking - Controls default rocking motion.
GRAPHICS.rocking Speed - Controls rocking or rotation speed.
GUI.auto Save Interval - Controls auto save period (minutes)
GUI.table Row Mark Colors - Controls colors used for marking tables.
GUI.workspaceTabStyle - Controls the style of ICM-object tabs created in the workspace panel of ICM GUI.
Movie.fade Nof Frames - Controls number of frames for the fade out option in screenshot movie making.
Movie.quality - Controls the resoltuion of the movie
SEQUENCE.site Colors - Controls coloring of squence sites.
SLIDE.ignore Fog - Fog representations can be ignored in slide preparation if desired.
GRAPHICS.discrete Rainbow -
GRAPHICS.rainbow Bar Style - determines if and where the color bar will appear after a molecule is colored by an array.
GRAPHICS.rocking Range - real value of rocking range.
GUI.auto Save - auto save on or off
GUI.max Sequence Length - maximum sequence length displayed in ICM
GUI.workspace Folder Style - Workspace folder style.
MOVIE.frame Grab Mode - with screenshot movie making you can choose either fixed frame time or real time.
Movie.quality Auto - with screenshot movie making you can allow ICM to control the movie resolution.
SLIDE.ignore Background Color - Ignore background color when you are making a slide.
3.7.5 GUI Preferences |
To change GUI Preferences:
- Select File/Preferences.
- Choose the GUI tab.
Quality - controls the quality (density of graphical elements) of such representations as cpk, ball, stick, ribbon . Do not make it larger than about 20 or smaller than 1. We recommend to make this parameter at least 15 if you want to make a high quality image. You can also increase the number of image resolution by making the image window 2,3,4 times larger (in the example below it is 2 times larger) than the displayed window.
Wire Style - Four different wire styles are available.
Hydrogen Display - Select whether you always want all hydorgens displayed or just-polar hydrogens or no hydrogens at all.
Rainbow Scale - determines if and where the color bar will appear after a molecule is colored by an array. Coloring by an array is one of the options of the display and color commands.
1. = "left" <- default choice 2. = "right" 3. = "no text" 4. = "no bar"
Ball Ratio - The ratio of ball and stick radii. This ratio is applied when the styles are switched to xstick from the GUI xstick toolbar.
Selection Style - Change the graphical display of your selections. Default is a green cross.
Clash Threshold - a clash is defined as an interatomic distance less than a sum of van der Waals radii of two atoms of interest multiplied by the clashThreshold parameter. For hydrogen bonded atoms, the distance threshold is additionally reduced by 20% . Default = 0.82
DotSurfaceRadiusIncrement - adius of a probe sphere used to display a dotted surface of a molecule. All van der Waals radii are expanded by this value. vwExpand=0 corresponds to the CPK surface, vwExpand=1.4 corresponds to the water-accessible surface. Be aware of the difference between the waterRadius and vwExpand parameters: waterRadius is used in
- show energy "sf"
- show [area|volume] skin
- display skin while vwExpand is used in
- show [area|volume] surface
- display surface
H Bond Style - How do you wish your H-Bonds to be displayed by default? Dashes, Bond Length, Bond Lenght and Angle.
grobLineWidth - relative width of displayed lines of 3D meshes ( grobs ). Also affects the interatomic distance display.
general line with - the real width of lines used to display the wire representation of chemical bonds. See also IMAGE.lineWidth parameter which controls line thickness in molecular images generated by the write postscript command, and the PLOT.lineWidth which controls the width for the plot command. Default (1.0)
single atom as - display style of isolated atoms in the wire mode.
1. "tetrahedron" 2. "cross" 3. "dot" The size of the first two representation is controlled by the GRAPHICS.ballRadius parameter and the line width (especially important for the "dot" style) is controlled by the lineWidth parameter.
xstickhetatomzZoom - The default ball and stick radii of a ligand can be different. This makes a better ligand view since the ligand stands out from the surrounding protein atoms.
solid shine style - choose either white or color
Stick Radius - radius (in Angstroms) of a cylinder displayed as a part of stick or xstick graphical representation of a molecule. Individual (residue-wide) control of stick radii.
Stereo Mode - Select a default stereo mode
Display Style - A default display style can be chosen using a combination of styles.
Water Radius - radius of water sphere which is used to calculate an analytical molecular surface (referred to as skin) as well as the solvent-accessible surface (centers of water spheres).
clashWidth - relative width of a displayed clash.
hbondWidth - relative width of hydrogen bond display
mapLineWidth - relative width of lines and dots of a displayed map.
3.7.6 Image Preferences |
To change Image Preferences:
- Select File/Preferences.
- Choose the Image tab.
IMAGE.color - logical to save color or black_and_white ('bw') images.
IMAGE.gammaCorrection - real variable to to lighten or darken the image by changing the gamma parameter. A gamma value that is greater than 1.0 will lighten the printed picture, while a gamma value that is less that 1.0 will darken it.
IMAGE.lineWidth - this real parameter specifies the default line width for the postscript lines.
IMAGE.orientation - image orientation.
IMAGE.previewer - a string parameter to specify the external filter which creates a rough binary (pixmap) postscript preview and adds it to the header of the ICM-generated high resolution bitmap or vectorized postscript files saved by the write image postscript, and write postscript , respectively .
IMAGE.print - unix command for printer.
IMAGE.scale - real variable. If non zero, controls the image scale with respect to the screen image size.
IMAGE.stereoBase - real variable to define the stereo base (separation between two stereo panels) in the write image postscript and write postscript command.
IMAGE.writeScale - an integer parameter used to increase the image resolution in the Quick Image Write tool.
IMAGE.bondLength2D - real length of a chemical bond (in inches) in chemical 2D drawings upon the Copy Image command.
IMAGE.compress - logical to toggle simple lossless compression, standard for .tif files. This compression is required to be implemented in all TIFF-reading programs.
IMAGE.generateAlpha - logical to toggle generation of the alpha (opacity) channel for the SGI rgb, tif and png image files to make the pixels of the background color transparent.
IMAGE.lineWidth2D - integer thickness of bonds in chemical 2D drawing upon the Copy Image command. This is useful for cutting and pasting from ICM to external documnents.
IMAGE.paper Size - specify paper size.
IMAGE.previewResolution - integer resolution of the rough bitmap preview added to the vectorized postscript file in lines per inch.
IMAGE.printerDPI - this integer parameter the printer resolution in Dot Per Inch (DPI). Important for the write image postscript command.
IMAGE.stereoAngle - real variable to define stereo angle (relative rotation of two stereo images) in the write image postscript and write postscript command.
IMAGE.stereoText - logical to make text labels for only one panel or both panels of the stereo diagram.
3.7.7 Label Preferences |
To change Label Preferences:
- Select File/Preferences.
- Choose the Labels tab.
atomLabelStyle style of atom labels invoked by clicking on an the atom label button.
GRAPHICS.displayLineLabels - enables/disables the display of edge lengths (inter-point distances) of a grob generated with the Grob( "distance" .. ) function.
GRAPHICS.font Line Spacing - Change the spacing between lines in labels.
GRAPHICS.resLabelDrag - if yes, enables dragging of the displayed residue labels with the middle mouse button.
GRAPHICS. site Arrow - Highlight sites with an arrow yes or no.
Show Res Code In Selection - When you make a selection the icm selection language will be displayed when you right click on the selection.
Res Label Style - Default residue label style.
SITE.label Style - Default label sites style.
Var Label Style - Default label variable style.
GRAPHICS.atomLabelShift - a non-negative integer number of spaces preceding an atom label. This parameter is useful for displaying labels next to a solid representation,
GRAPHICS.fontColor - set font color
GRAPHICS.font Scale - set font size
GRAPHICS.site Label Shift - GRAPHICS.resLabelShift a non-negative integer number of spaces preceding a site label.
GRAPHICS. site Label Drag - if yes, enables dragging of the displayed site labels with the middle mouse button.
Res Label Shift - a non-negative integer number of spaces preceding a residue label. This parameter is useful for displaying residue labels next to a solid
SITE.labelOffset - (default 5. A) the real offset of the site label with respect to the residue label atom.
SITE.wrap Comment - Number of characters per comment line.
3.7.8 Plot Preferences |
To change Plot Preferences:
- Select File/Preferences.
- Choose the Plot tab.
PLOT.color - logical to generate a color plot. Usually it does not make sense to switch it off because your b/w printer will interpret the color postscript just fine anyway.
PLOT.draw Tics logical yes or no
PLOT.fontSize real font size. Any reasonable number from 3. (1 mm, use a magnifying glass then) to 96.
PLOT.lineWidth - real line width for graphs (not the frame and tics)
PLOT.markSize - real mark size in points. Allowed mark types: line, cross, square, triangle, diamond, circle, star, dstar, bar, dot, SQUARE, TRIANGLE, DIAMOND, CIRCLE, STAR, DSTAR, BAR. Uppercase words indicate filled marks.
PLOT.paper Size - preference to specify plor paper size
PLOT.rainbowStyle - preference defining the color spectrum used by the plot area command.
PLOT.Yratio - real aspect ratio of the ICM plot frame. Using link option of the plot command is equivalent to setting this variable to 1.0. If PLOT.Yratio is set to 0. , the ratio will be set automatically to fill out the available box optimally.
[PLOT.date} - display date on plot
PLOT.font - preference for the title/legend font.
PLOT.labelFont - preference for the data point label font.
PLOT.logo - logical switch for the ICM-logo on the plot.
PLOT.orientation - preference for the plot orientation.
PLOT.previewer - command to local ps viewer
PLOT.seriesLabels - preference to indicate position of a series/color legend inside the plot frame.
3.7.9 Ribbon Preferences |
To change Ribbon Preferences:
- Select File/Preferences.
- Choose the Ribbon tab.
Combo Display Style - select ribbon-cpk, atoms, ribbon-ligand, chemical
GRAPHICS.dnaRibbonRatio - real ratio of depth to width for the DNA ribbon .
GRAPHICS.dnaRibbonWorm - logical which, if yes, makes the DNA backbone ribbon round, rather than rectangular. Default: no
GRAPHICS.dnaWormRadius - real radius of the worm representing bases in DNA ribbon .
GRAPHICS.ribbonWidth - real width of the protein ribbon .
GRAPHICS.wormRadius - radius of coiled segments (i.e. those where the secondary structure is marked as "_") of a polypeptide chain in ribbon representation. Default (0.3).
Ribbon Style - specifies default style when ribbon is displayed.
GRAPHICS.dnaBallRadius - DNA bases in ribbon representation are shown as balls controlled by this real parameter.
GRAPHICS.dnaRibbonWidth - real width (in Angstroms) of the DNA ribbon .
GRAPHICS.dnaStickRadius - real radius of the sticks representing bases in DNA ribbon .
GRAPHICS.ribbonRatio - real ratio of depth to width for the protein ribbon .
GRAPHICS.ribbonWorm - logical parameter, if yes, makes the ribbon round, rather than rectangular.
ribbonColorStyle -
- sets the ribbon coloring scheme. 1 = "type" default. colors by secondary structure type or explicit color 2 = "NtoC" colors each chain gradually blue-to-red from N- to C- (or from 5' to 3' for DNA) 3 = "alignment" if there is an alignment linked to a protein, color gapped backbone regions gray 4 = "reliability" 3D gaussian averaging with selectSphereRadius of alignment strength in space If ribbonColorStyle equals to 4, the conserved areas will be colored blue, while the most divergent will be red, and the intermediate conservation areas will be colored white. Example:
3.7.10 Shell Preferences |
To change Shell Preferences:
- Select File/Preferences.
- Choose the Shell tab.
Clash Threshold - a clash is defined as an interatomic distance less than a sum of van der Waals radii of two atoms of interest multiplied by the clashThreshold parameter.
Map Sigma Level - (in Rmsd values over the mean value). Margin value used for making graphical objects contouring the 3D density map .
Mnconf - maximal number of conformations in the conformational stack . The stack stops growing after this number is achieved and starts replacing representative conformations with higher energy values by new conformations with superior energies, if the latter are found.
Icm Prompt - defines the ICM-prompt string.
Select Min Grad - default minimal gradient vector length for gradient atom selection ( a_//G). This parameter is also used by the montecarlo fast command, which requires a value of 2. to 10. for optimal performance.
Map Atom Margin - Margin in Angstoms around selected atoms. The margin is added to the positional boundaries to define a submap index box in the Map ( map_source , as_ ) function.
maxColorPotential - local electrostatic potential in kcal/e.u.charge units at which the surface element is colored by extreme red or extreme blue. All higher values will have the same color. This absolute scaling is convenient to develop a feeling of electrostatic properties of molecular surfaces.
mnSolutions - this parameter limits the number of hits retained by the program after a search.
Real Format - format of real numbers
Water Radius - radius of water sphere which is used to calculate an analytical molecular surface
3.7.11 System Preferences |
To change System Preferences:
- Select File/Preferences.
- Choose the System tab.
FTP.createFile -
FTP.proxy - string path to the proxy server for connections through firewall. Default: "" (empty string).
GUI.max Nof Recent Files - maximum number of recent files stored.
GUI.splash Screen Image - path to splash image displayed on startup
HTTP. support Cookies - http support cookies yes or no
HTTP.user Agent - client application used within a particular network protocol for www
Beep - warning beep yes or no
Max File Size Mb - Maximu file size in MegaBytes that can be loaded into ICM.
USER.friends
USER.organization
FTP.keep File - (default no ). If yes, the temporary file is kept in the s_tempDir directory. Otherwise the file is deleted.
GUI.enumberation Memory Limit - memory limit for enumeration operations.
GUI.splash Screen Delay
HTTP.proxy - string for HTTP server for connection through firewall
HTTP.timeout - timeout in seconds
Http Read Style icm or lynx
Force Auto Bond Typing - yes/no
USER.email, USER.full Name, USER.phone
| Prev Tags | Home Up | Next GUI Basics |