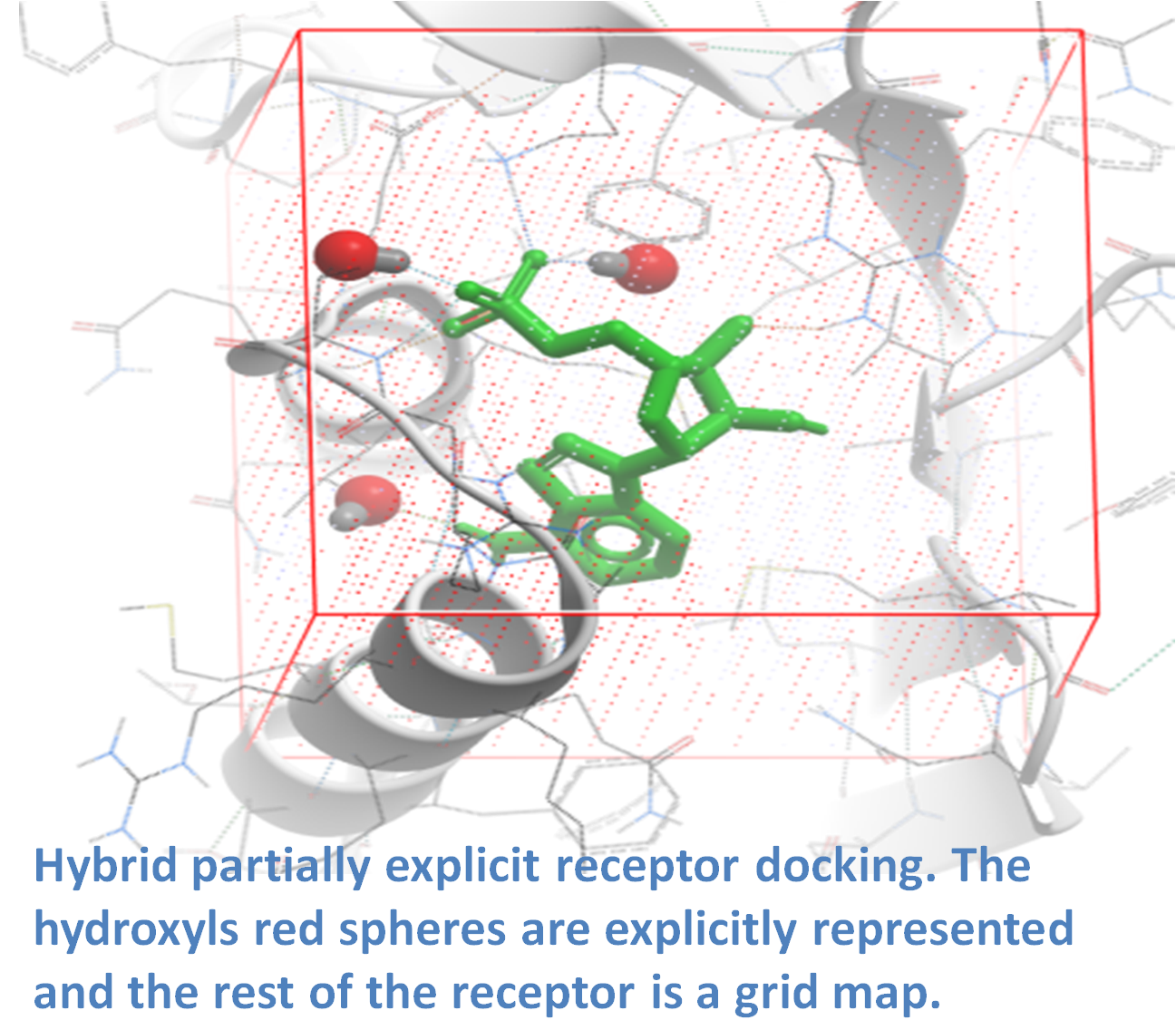
 ICM contains a selection of tools to account for pocket flexibility (induced fit) in docking and virtual screening. The importance of considering flexibility in proteins is well understood (1-3) and is an important consideration when undertaking structure-based drug design. There are three main approaches in ICM for incorporating induced fit:
ICM contains a selection of tools to account for pocket flexibility (induced fit) in docking and virtual screening. The importance of considering flexibility in proteins is well understood (1-3) and is an important consideration when undertaking structure-based drug design. There are three main approaches in ICM for incorporating induced fit:
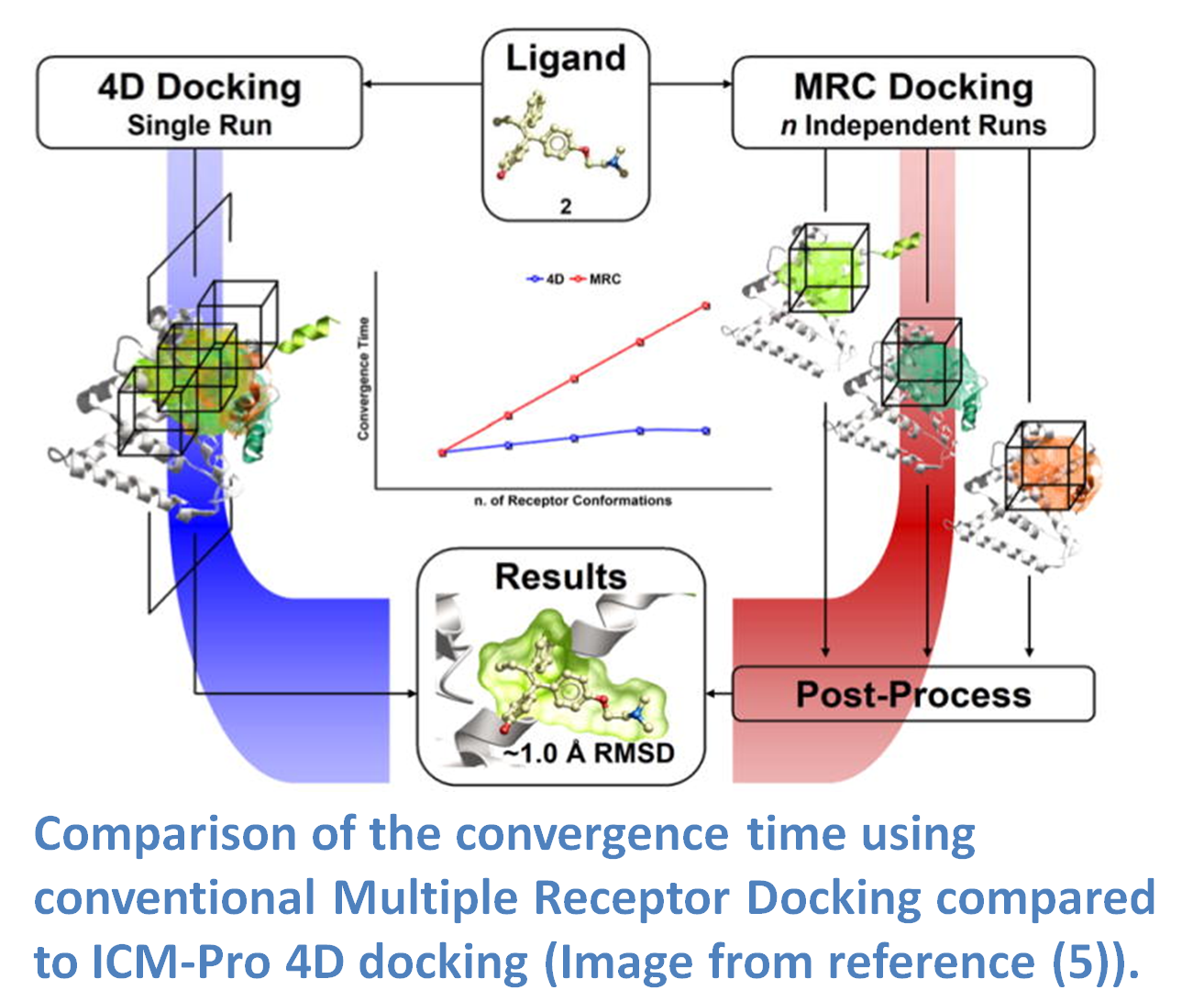 If you have multiple experimental X-Ray structures of your receptor then you are in a good position to start 4D docking. However, the most common starting point is a single crystal structure or homology model and so a method to sample pocket flexibility is needed. ICM contains methods for generating an ensemble of pocket structures for 4D docking these include: Fumigation, Normal Modes, and Ligand Guided Modeling.
If you have multiple experimental X-Ray structures of your receptor then you are in a good position to start 4D docking. However, the most common starting point is a single crystal structure or homology model and so a method to sample pocket flexibility is needed. ICM contains methods for generating an ensemble of pocket structures for 4D docking these include: Fumigation, Normal Modes, and Ligand Guided Modeling.
4D docking allows the ligand to sample an ensemble of receptor conformations in a single docking run. The potential maps are generated for all the conformations and stored in a single multi-dimensional map file. The method works by generating grid potential maps for each receptor and they are stored in a single data structure referred to as a 4D grid. The docking procedure then runs using MolSoft's Biased Probability Monte Carlo (BPMC) method and the ligand samples the 3D Cartesian coordinates of the grid sampling nodes and a fourth coordinate (indexed receptor conformations) by a special type of random move. It has been found that the convergence time for 4D docking is comparable to that of regular docking and is significantly faster than conventional multiple receptor docking (image - right). The method was benchmarked on 99 therapeutically relevant proteins and 300 diverse ligands and was found to have an accuracy of ~80% in ligand pose prediction (5).
Lead Discovery using Ligand Guided Modeling. Ligand guided modeling was applied to the human Androgen Receptor (AR) and two models were chosen for virtual screening. More than 2000 marketed drugs were screened to the models and 11 of the top scoring compounds were tested experimentally. Four of the compounds were antipsychotic drugs and inhibited AR at 300-500nM. In 2008, the methods was used for the challenging case of Melanin Concentrating hormone which is a GPCR implicated in obesity (8). 800 models were constructed using known MCH antagonists, they were then filtered down to a representative set of structures for virtual screening. A database of >187K compounds were docked to the models and 281 compounds were tested experimentally resulting in 6 active compounds which represents a >10-fold enrichment rate compared to traditional HTS. The method was also applied to the Adenosine A2A GPCR where 56 compounds sent for experimental testing in functional assays. 23 compounds were identified with affinity <10 µM and 11 of those had had sub-µM affinities and 2 had affinities <60nM representing a diverse and novel set of antagonist scaffolds.
Accurate Models of GPCR Agonist Pockets using Ligand Guided Modeling. Ligand guided modeling was also used to accurately predict the agonist form of a GPCR. The agonist models of Beta 2 Adrenergic receptor and Adenosine A2A generated with ICM were published (10,11) and in 2010 the crystal structure was published (12). A comparison between the ICM models and the atomic crystal structure showed that the agonist binding pose of the agonist differed by only 0.8≈. This comparison was recently summarized in TIPS (13) along with another accurate prediction of the Adenosine A2A agonist receptor structure.
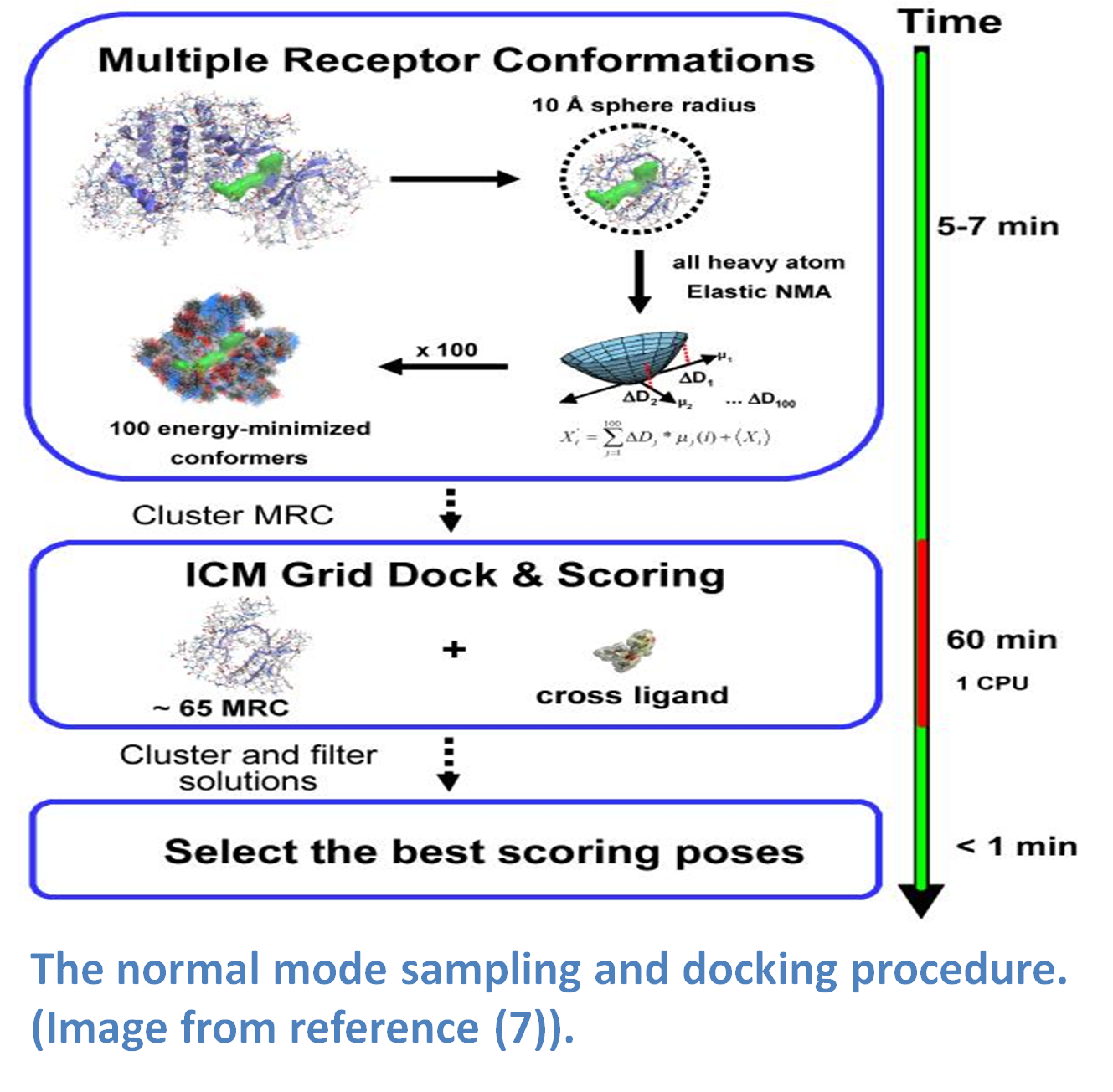 Normal modes is a method that provides a spring-like representation of the pocket backbone atoms allowing a wide conformational space to be sampled (5,14). The method (image - right) does not require a priori knowledge of the region that is being sampled and has performed well in cross docking benchmarks (5). The approach uses a Hookean potential to describe the interaction energy between two atoms.
Normal modes is a method that provides a spring-like representation of the pocket backbone atoms allowing a wide conformational space to be sampled (5,14). The method (image - right) does not require a priori knowledge of the region that is being sampled and has performed well in cross docking benchmarks (5). The approach uses a Hookean potential to describe the interaction energy between two atoms.
A heavy atom ICM Elastic Network NM modeling approach was successfully used in the 2008 blind G-Protein Coupled Receptor (GPCR) modeling competition. The method yielded the best model in the competition for the number of ligand-receptor contacts for the Adenosine A2a receptor (15,16).
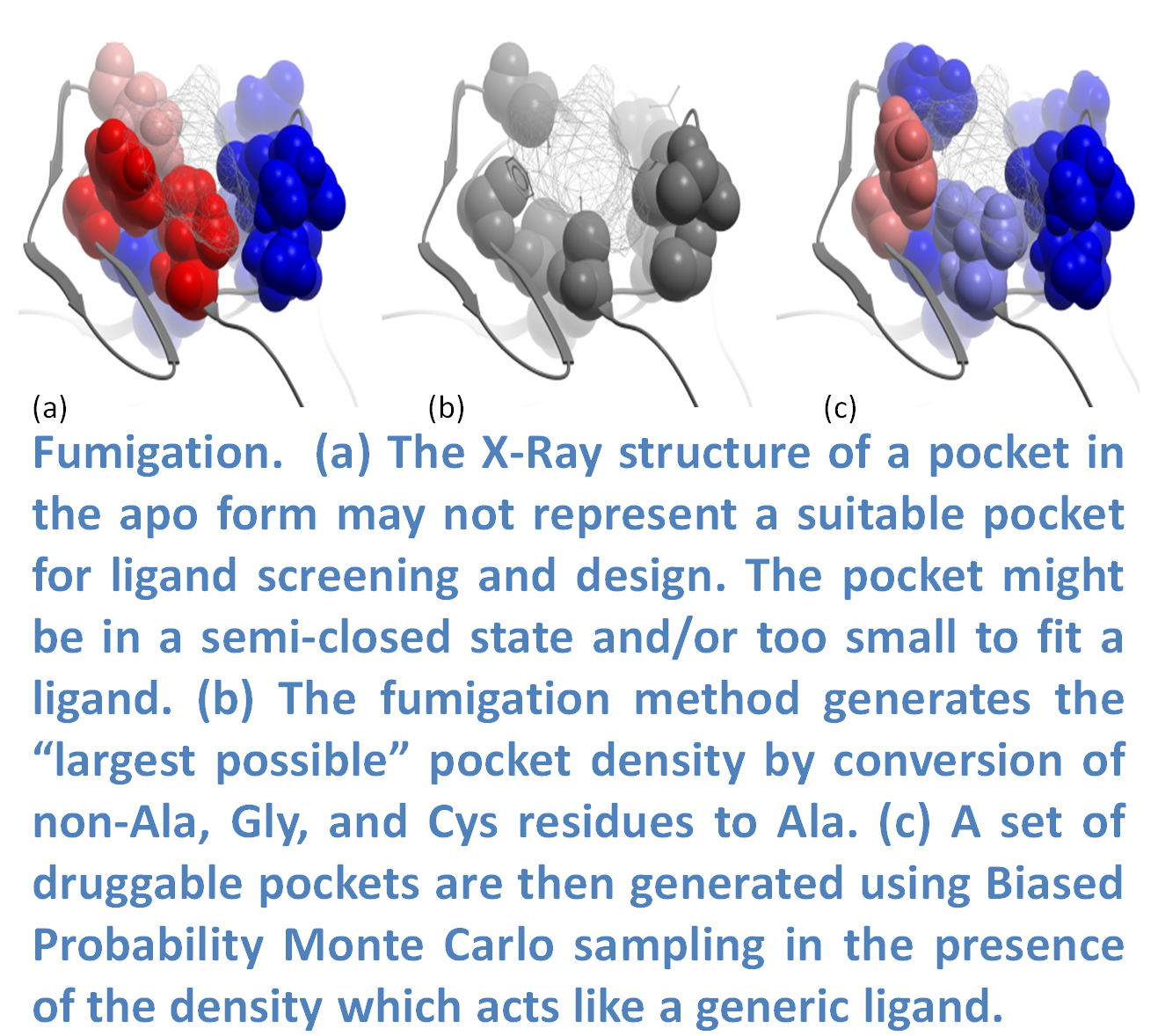 In many cases the ligand binding pocket observed in a crystal structure does not represent the ligand-compatible conformation. Fumigation is a modeling method developed by Ruben Abagyan.s lab at UCSD and first reported at the Protein Society Meeting in 2008 (17). The method generates a set of druggable conformations of apo small molecule ligand binding pockets. The technique is based on sampling the torsion angles of the pocket side-chains in the presence of a repulsive density which represents a generic ligand. The procedure is summarized in the image (left) and described below. The first step is to generate the largest possible state of a pocket by simultaneous conversion of the pocket residues to Alanine (Ala, Gly, and Cys residues are excluded). Next, an atom density grid map for the "shaved out" pocket is generated and smoothed via spatial averaging of the map. The spatial averaging of the map is repeated to fill the cavities of the original protein and then the difference between the smoothed and original maps is calculated. The generated density is used as a penalty term in the energy calculations when the internal variables of the pocket residues are sampled in ICM using BPMC (18). The procedure results in an ensemble of pockets which can be used for virtual ligand screening.
Recent published applications of the Fumigation method include a determination of the binding mode19 and design20 of Serotonin Receptor inhibitors and the discovery of novel cyclic guanosine monophosphate (cGMP) efflux inhibitors21.
In many cases the ligand binding pocket observed in a crystal structure does not represent the ligand-compatible conformation. Fumigation is a modeling method developed by Ruben Abagyan.s lab at UCSD and first reported at the Protein Society Meeting in 2008 (17). The method generates a set of druggable conformations of apo small molecule ligand binding pockets. The technique is based on sampling the torsion angles of the pocket side-chains in the presence of a repulsive density which represents a generic ligand. The procedure is summarized in the image (left) and described below. The first step is to generate the largest possible state of a pocket by simultaneous conversion of the pocket residues to Alanine (Ala, Gly, and Cys residues are excluded). Next, an atom density grid map for the "shaved out" pocket is generated and smoothed via spatial averaging of the map. The spatial averaging of the map is repeated to fill the cavities of the original protein and then the difference between the smoothed and original maps is calculated. The generated density is used as a penalty term in the energy calculations when the internal variables of the pocket residues are sampled in ICM using BPMC (18). The procedure results in an ensemble of pockets which can be used for virtual ligand screening.
Recent published applications of the Fumigation method include a determination of the binding mode19 and design20 of Serotonin Receptor inhibitors and the discovery of novel cyclic guanosine monophosphate (cGMP) efflux inhibitors21.
Fumigation of a Protein-Protein Interaction Site. The Fumigation method was first used for the discovery of protein-protein interaction inhibitors (5.4nM potency) in the Protein Kinase CK2a-CK2b interface (17). 2000 pockets were generated and clustered and a subset was chosen to be used in ICM virtual ligand screening. 100 compounds were tested experimentally for activity and 14 were found to inhibit the interaction from 25-60% in a dose dependent manner.
2. Orry, A. J. W. & Abagyan, R. Preparation and refinement of model protein-ligand complexes. Methods Mol. Biol. 857, 351.373 (2012).
3. Totrov, M. & Abagyan, R. Flexible ligand docking to multiple receptor conformations: a practical alternative. Curr. Opin. Struct. Biol. 18, 178.184 (2008).
4. Kufareva, I., Ilatovskiy, A. V. & Abagyan, R. Pocketome: an encyclopedia of small-molecule binding sites in 4D. Nucleic Acids Res. 40, D535.540 (2012).
5. Rueda, M., Bottegoni, G. & Abagyan, R. Consistent improvement of cross-docking results using binding site ensembles generated with elastic network normal modes. J Chem Inf Model 49, 716.725 (2009).
6. Bottegoni, G., Rocchia, W., Rueda, M., Abagyan, R. & Cavalli, A. Systematic exploitation of multiple receptor conformations for virtual ligand screening. PLoS ONE 6, e18845 (2011).
7. Katritch, V., Rueda, M. & Abagyan, R. Ligand-guided receptor optimization. Methods Mol. Biol. 857, 189.205 (2012).
8. Cavasotto, C. N. et al. Discovery of novel chemotypes to a G-protein-coupled receptor through ligand-steered homology modeling and structure-based virtual screening. J. Med. Chem. 51, 581.588 (2008).
9. Bisson, W. H. et al. Discovery of antiandrogen activity of nonsteroidal scaffolds of marketed drugs. Proc. Natl. Acad. Sci. U.S.A. 104, 11927.11932 (2007).
10. Katritch, V. et al. Analysis of full and partial agonists binding to beta2-adrenergic receptor suggests a role of transmembrane helix V in agonist-specific conformational changes. J. Mol. Recognit. 22, 307.318 (2009).
11. Reynolds, K. A., Katritch, V. & Abagyan, R. Identifying conformational changes of the beta(2) adrenoceptor that enable accurate prediction of ligand/receptor interactions and screening for GPCR modulators. J. Comput. Aided Mol. Des. 23, 273.288 (2009).
12. Wacker, D. et al. Conserved binding mode of human beta2 adrenergic receptor inverse agonists and antagonist revealed by X-ray crystallography. J. Am. Chem. Soc. 132, 11443.11445 (2010).
13. Katritch, V. & Abagyan, R. GPCR agonist binding revealed by modeling and crystallography. Trends Pharmacol. Sci. 32, 637.643 (2011).
14. Cavasotto, C. N., Kovacs, J. A. & Abagyan, R. A. Representing receptor flexibility in ligand docking through relevant normal modes. J. Am. Chem. Soc. 127, 9632.9640 (2005).
15. Michino, M. et al. Community-wide assessment of GPCR structure modelling and ligand docking: GPCR Dock 2008. Nat Rev Drug Discov 8, 455.463 (2009).
16. Katritch, V., Rueda, M., Lam, P. C.-H., Yeager, M. & Abagyan, R. GPCR 3D homology models for ligand screening: lessons learned from blind predictions of adenosine A2a receptor complex. Proteins 78, 197.211 (2010).
17. Kufareva,, I., Laudet,, B., Cochet,, C. & Abagyan, R. Structure-based discovery of small molecules that modulate kinase activity by disrupting the subunit interaction: application to CK2. Protein Science 17 Supplementary 1, 265
18. Abagyan, R. & Totrov, M. Biased probability Monte Carlo conformational searches and electrostatic calculations for peptides and proteins. J. Mol. Biol. 235, 983.1002 (1994).
19. Gabrielsen, M. et al. Molecular mechanism of serotonin transporter inhibition elucidated by a new flexible docking protocol. Eur J Med Chem 47, 24.37 (2012).
20. Jaro.czyk, M. et al. Synthesis, in vitro binding studies and docking of long-chain arylpiperazine nitroquipazine analogues, as potential serotonin transporter inhibitors. Eur J Med Chem 49, 200.210 (2012).
21. Sager, G. et al. Novel cGMP Efflux Inhibitors Identified by Virtual Ligand Screening (VLS) and Confirmed by Experimental Studies. J. Med. Chem. 55, 3049.3057 (2012).
22. Bottegoni, G., Kufareva, I., Totrov, M. & Abagyan, R. A new method for ligand docking to flexible receptors by dual alanine scanning and refinement (SCARE). J. Comput. Aided Mol. Des 22, 311.325 (2008).
© 2026 All Rights Reserved MolSoft LLC Terms of Use | Privacy Policy



