| Prev | ICM User's Guide 14 Molecular Dynamics | Next |
Molecular Dynamics (MD) in ICM uses the functions in OpenMM - if you use this feature please cite OpenMM.
To run an MD simulations using Amber force field ff14SB:
- Convert your protein to an ICM object.
- MolMechanics/Run MD Simulation
- Use the drop down box to select the protein.
- Select whether or not you wish to solvate the protein.
- Select the length of time for the simulation in ns.
- Click OK.
To add restraints:
- Make a copy of the protein you wish to simulate - right click on the name of the protein in the ICM workspace (left hand side) and choose the option 'clone'.
- Select the region of the cloned molecule you wish to restrain and convert that selection to an 'orange' selection as described here.
- Go to MolMechaics/Run MD Simulation as described above and choose the selection in the Restrain dialog box.
- Click OK to run the simulation.
Results:
The conformation snapshots are stored at regular time intervals during the MD simulation and are embedded in the ICM object as a stack.
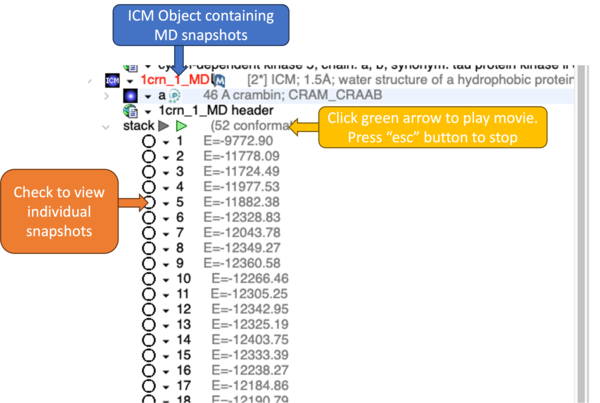
Results Analysis:
Right click on the stack of MD snapshots in the ICM workspace and choose Stack Calculations. In this dialog box you can analyze
- Distance - Select a pair of atoms or use green and orange selections for closest distance. A table and plots of the distance will be displayed.
- RMSD to Reference - Read in a separate object as a reference and make the selection you want to analyze. You can choose to analyze the structures 'static' or superimpose. A table and plot of the RMSDs are displayed. This is a useful tool for analysis of a Molecular Dynamics run. Your x-axis will be the time in ns / number of conformations so if you chose 1ns and 50 conformations (snapshots) then each point is 1ns/50 = 0.02 ns
- RMSF - Root mean square fluctuation indicates positional differences in structure during a simulation. There is a Static and Superimpose option - the superimpose option follows protocol described here. You can provide two selections: for superposition using orange selection and regular "green" selection for RMSF calculation. By default it (if no selections are provided) backbone from all residues will be used.
- Ligand Contacts - This method uses our Interaction Lists macro from our docking hitlist. Select the ligand and then the macro will be called for each MD snapshot in the stack and calculates number of: hydrophobic(apol), hydrogen bond don/acc (hba/hbd) and positive/negative. Individual residues can be included if you check this opti
- Torsion - Select a pair of atoms on a rotatable bond to calculate the torsion angle.
- Area - use green and orange selections to calculate differences in area
- RTCNN - use green and orange selections for receptor/ligand respectively to calculate RTCNN docking score.
- ClusterTree - make a selection of atoms to compare and then a cluster tree will be displayed. You can then 'select-tree{select} a diverse set of conformations in the tree.
| Prev RIDE GPU Benchmark | Home Up | Next MolScreen |