 MERSi is a new algorithm available in ICM-Pro that allows you to predict the effect of a mutation upon protein stability and protein-protein binding free energy.
MERSi is a new algorithm available in ICM-Pro that allows you to predict the effect of a mutation upon protein stability and protein-protein binding free energy.
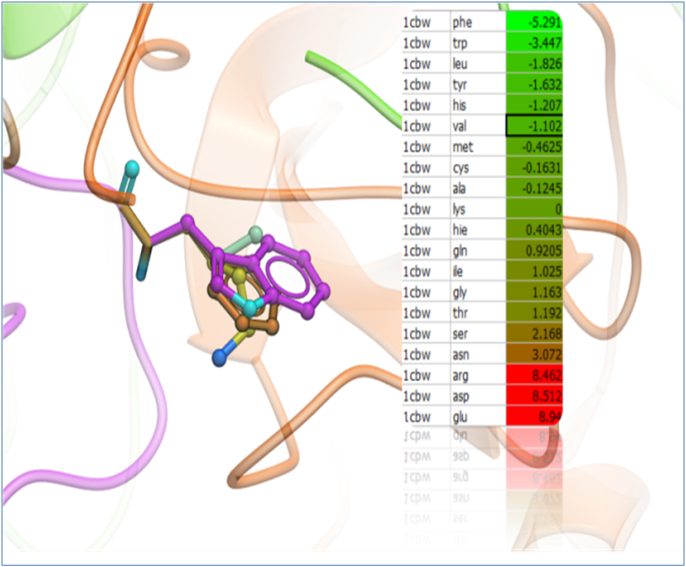
Predict Change in Binding Free Energy upon Mutation The binding free energy change ΔΔGbind, is computed as a difference between the binding free energy of mutant and wild type complexes. To account for structure relaxation upon mutation, side chain re-packing in the vicinity of the mutated residue by a Monte Carlo simulations carried out. "Scan Protein Interface" option allows to mutate all residues (one by one) of the Interacting Part in close contact with the second part of the complex.
Separate mutation prediction protocols are available for Protein-protein, Protein-peptide and Protein-ligand Binding.
Predicting Change in Protein Stability upon Mutation The stability change is calculated as a weighted sum of a complete set of physically meaningful free-energy contributions, including van der Waals attractions, repulsions, electrostatics, hydrogen bonding, and solvation term. Residue-specific constants that account for the free energies of the unfolded and misfolded states were derived empirically using a large set of experimental data. Mutation of a given residue is followed by Monte Carlo simulations with flexible side chains for the mutated residue and its neighboring residues. Backbone flexibility is also considered for special cases such as proline and disulfide-forming/breaking cystein mutations.
Predicting the Effect of Mutations in SARS-COV-2 The Structural Genomics Consortium have been using ICM to calculate ΔΔG of binding inhibitor bidning in catalytic pocket of SARS-CoV-2 Papain-like protease (PLPro). You can read more here and here in the SGC's open lab notebook.
Please Cite Schapira M, Totrov M, Abagyan R. Prediction of the binding energy for small molecules, peptides and proteins. J Mol Recognit. 1999 May-Jun;12(3):177-90. PMID: 10398408 The protocol and parameterization has been re-optimized since this publication, but it is the same method.
© 2026 All Rights Reserved MolSoft LLC Terms of Use | Privacy Policy



