State of the Art Physics-Based and AI Desktop Modeling Software
Platforms Available : Windows, Linux, Mac OSX
ICM-Pro add-ons : ICM-Homology, ICM-Chemist-Pro and ICM-VLS
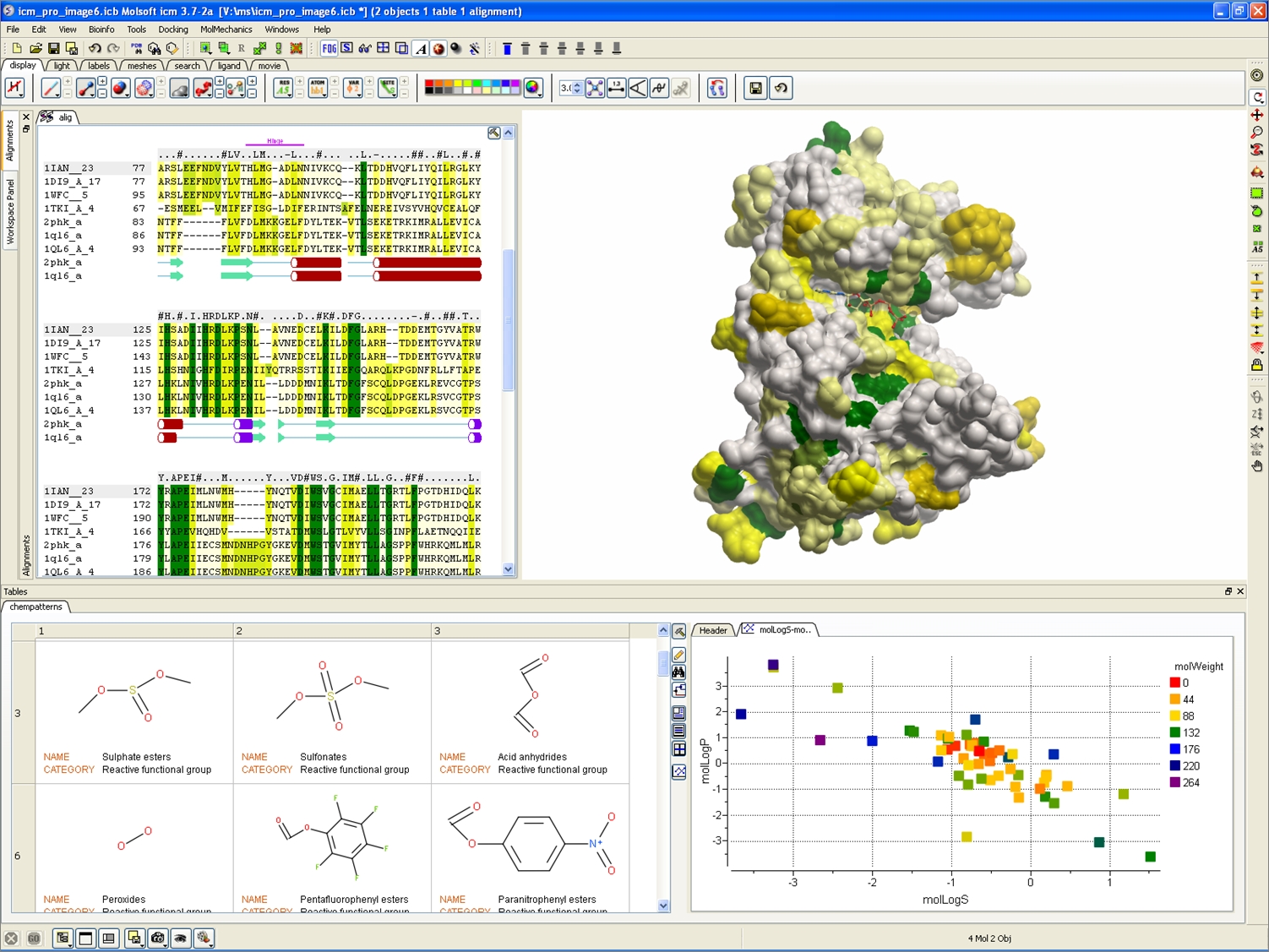 | ICM-Pro empowers a biologist or chemist by providing a high quality protein structure analysis, modeling, and docking desktop software environment. You have direct access to sequence and structural databases which allows you to: analyze sequences and alignments, inspect protein structure, study pockets and bound ligands and drugs, create surfaces, calculate electrostatics, make mutations, predict ligand binding sites, predict protein-protein interaction sites, perform small molecule and protein-protein docking and design ligands using the Interactive Ligand Editor. |
 |
Small Molecule Docking. ICM-Pro provides a unique set of tools for the modeling of protein/ligand interactions. ICM-Pro performs fast and accurate docking of fully continuously flexible small molecule ligands to a protein represented by grid interaction potentials. There are also inbuilt procedures to account for induced fit which include multiple receptor docking (4D docking) and explicit receptor docking. Read more... |
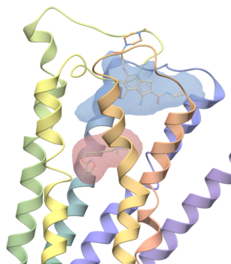 | ICM Pocket Finder. ICM Pocket Finder is a method which will identify ligand binding sites in any protein, DNA or RNA structure. Protein-ligand binding sites are identified based on the grid potential map of van der Waals interaction of the receptor and the surfaces are contoured. Physical properties of the pockets are then calculated and tabulated and a drug-likeness score is provided. Read more... |
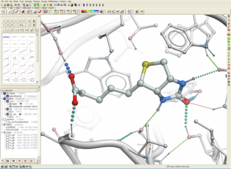 | ICM 3D Ligand Editor. Developed by MolSoft in Collaboration with Medicinal Chemists at Novartis. The 3D Ligand Editor allows you to interactively edit a chemical inside a receptor binding pocket. Modify atoms and groups and see the effect of the changes on ligand binding energy and score. A ligand can be modified in 2D or 3D and the effects of the modification can be seen on the binding energy to the receptor. For example a substituent can be changed with a single click on the screen and a calculation of the ligand binding score is made on the fly. The changes are stored and full undo and redo options are available and if the chemist likes the change they can tag and save the ligand in a chemical spreadsheet. Predictions are powered by MolSoft's high accurate docking ICM docking software. Read more... |
 |
Molecular Dynamics. MolSoft ICM-Pro includes built-in support for OpenMM, a powerful GPU-accelerated molecular dynamics (MD) engine.
With optimized GPU code, ICM-Pro enables fast MD simulations-achieving 3–12 ps/s (or 0.2–1.0 μs/day) on a cost-effective workstation GPU (e.g., a standard ‘gamer’ box). ICM-Pro features a direct binary interface to OpenMM, providing:
|
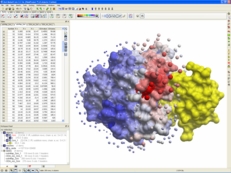
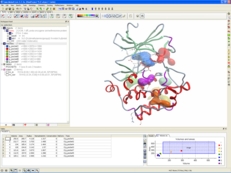 | Protein Structure Analysis. ICM-Pro provides a direct link to the Protein Data Bank (PDB). Once you have downloaded a structure you can analyse the structure - build Ramachandran plots, superimpose multiple structures, analyse distances and angles, calculate contact and surface areas, display hydrogen bonds, build electrostatic surfaces and calculate ligand binding pockets. Read more... |
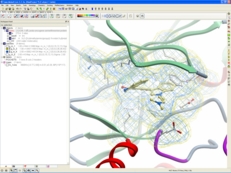 | Crystallographic Analysis Tools. The key to understanding a protein structure is to fully evaluate the underlying crystallographic information contained within a PDB file. For example, it is important to understand the full biological unit of a protein to identify if crystal-crystal contacts have influenced the structure or you may want to contour the electron density to see how much of a ligand was seen by the crystallographer in the active site. Read more... |
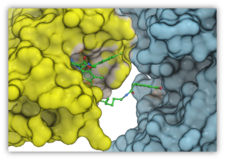
 | Protein-Protein Docking. ICM-Pro contains a well validated and successful protein-protein docking algorithm. ICM-Pro has consistently performed very well at the worldwide CAPRI protein-protein docking competition. ICM-Pro also contains an algorithm to predict protein-protein docking interaction sites. Read more... |
 | PROTAC Modeling Automatically and accurately model the tripartite complex of target protein-PROTAC molecules-Ligase. Read more... |
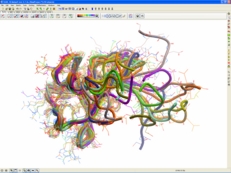 | Protein Structure Prediction. ICM-Pro has a good record in protein modeling. There are procedures which will regularize or build the backbone, shake up the side-chains and loops by global energy optimization. You can also perform simulations of small peptide folding. Read more... |
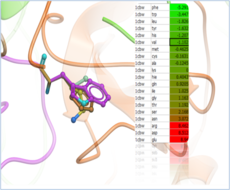 | Predict Effect of Mutation Predict the effect of a mutation on Protein Stability and Protein-Protein, Protein-Peptide and Protein-Ligand Binding. Read more... |
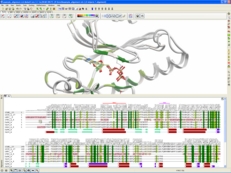 | Bioinformatics Tools. The ICM-Pro package contains all the tools in ICM-Bio which allows you to search a sequence database with high-quality global pairwise and multiple alignment algorithms and perform pattern, prosite and profile searches. Multiple sequence alignments are fast, the algorithm produces evolutionary trees, principal component views, annotation transfer from sequence to structures, threading and alignment visualization tools. Read more... |
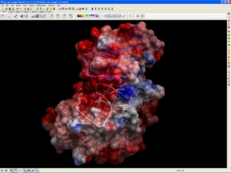 | Electrostatics. The Poisson equation for a molecule of any size can be efficiently solved using the boundary element algorithm that does not depend on any grid, and uses the exact analytical molecular surface as the boundary. ICM-REBEL calculates the accurate electrostatic potential of a molecule using boundary element algorithm and generates a 3D surface skin model colored by potential.Solves the Poisson equation for a molecule with exact positions of electric charges. Read more... |
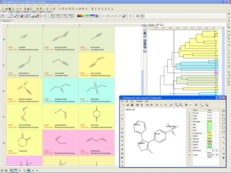 | Chemistry Tools. A variety of chemistry tools are available. Chemicals can be sketched in the ICM Molecular Editor and viewed in a chemical spreadsheet. Read more... |
 | Molecular Graphics.ICM-Pro contains all the tools in ICM-Browser-Pro a full and robust array of graphics tools all accessible from a GUI interface. Display your molecules in wire, CPK, ball&stick, worm, ribbon, accessible surface, transparent molecular surface, perspective, depth cueing, smooth and rugged solid surfaces. Use both hardware, side-by-side, and Anaglyph stereo. Read more... |
See the Minimum Recommended Hardware Specifications required to run the ICM-Pro software.
ICM Methods
Protein-Protein Docking
© 2026 All Rights Reserved MolSoft LLC Terms of Use | Privacy Policy



