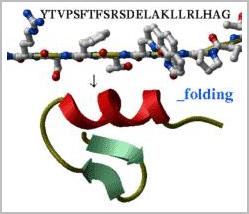
 Predicting low energy conformations for chemical compounds, peptides,
nucleic acids etc.: Take a peptide sequence and predict its three-dimensional
structure. Of course, the success is not guaranteed, especially if the peptide
is longer than about 25 residues but some preliminary tests are encouraging.
Evaluate local secondary structure preferences directly from the simulation.
Watch a movie with your peptide folding.
Predicting low energy conformations for chemical compounds, peptides,
nucleic acids etc.: Take a peptide sequence and predict its three-dimensional
structure. Of course, the success is not guaranteed, especially if the peptide
is longer than about 25 residues but some preliminary tests are encouraging.
Evaluate local secondary structure preferences directly from the simulation.
Watch a movie with your peptide folding.
Protein Modeling: ICM-Pro has a good record in protein modeling. There are procedures which will regularize or build the backbone, shake up the side-chains and loops by global energy optimization. You can also color the model by local reliability to identify the potentially wrong parts of the model. This does not include, however, the fast routine for building a complete model by homology with loops combined with the database search ( ICM-Homology is a separate add-on to ICM-Pro).
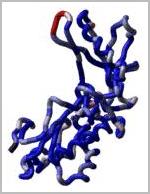 Loop modeling and protein design: ICM-Pro was used to design two new 7
residue loops and in both cases the designs were successful. Moreover, the
predicted conformations turned out to be exactly right (accuracy of 0.5A) after
the crystallographic structures of the designed proteins were determined by Rik
Wierenga and his coworkers.
Loop modeling and protein design: ICM-Pro was used to design two new 7
residue loops and in both cases the designs were successful. Moreover, the
predicted conformations turned out to be exactly right (accuracy of 0.5A) after
the crystallographic structures of the designed proteins were determined by Rik
Wierenga and his coworkers.
| Energy and Penalty Terms |
| A variety of energy and grid terms are available which are described in detail here |
| Set tethers restraints between atoms. |
| Delete tethers. |
| Set upper and lower boundaries of tethers. |
| Set tether weights. |
| Set tethers as a constant force. |
| Set distance restraints between atoms. |
| Minimization |
| Minimize Cartesian in Merck Molecular Force Field (MMFF) |
| Local minimization. |
| Minimize all atoms. |
| Minimize side chains. |
| Minimize hydrogens. |
| Minimize molecule position. |
| Adjust length of minimization. |
| Adjust a number of minimization parameters described here |
| Biased Probability Monte Carlo |
| Global optimization using the ICM Biased Probability Monte Carlo (BPMC) method (Abagyan et al 1994). |
| Adjust a number of BPMC parameters described here |
| Generate multiple receptor conformations (can be used in docking). |
| Optimize side chains. |
| Optimize ligand vicinity. |
| Optimize hydrogen bonds. |
| Loop Modeling |
| Sample loop conformations using BPMC |
| Fully-interactive table of loop conformations ranked by energy is displayed. |
| Click on the table to load loop conformation. |
| Force Fields |
| Select from 4 different force fields, ecepp, ecepp05, icmff and mmff |
| Set force field types and charges. |
| Regularization |
| Regularization options and features are described here |
| Edit Structure |
| Set bond type. |
| Set formal charge. |
| Set chirality. |
| Build hydrogens. |
| Set disulfide bonds. |
| Make bonds. |
| Make bonds by distance. |
| Delete bonds. |
| Set occupancy. |
| Set B-factor. |
| Make mutations and see the effect on energy of the structure or use for docking. |
| Modify group. |
| Randomize atoms. |
| Collection of Molecule Conformations(Stack). |
| View stack by loading a fully interactive clickable table. |
| Play all stack conformations in a loop (movie-like). |
| Add an additional conformation to an existing stack. |
| Store stack in an object. |
| Delete stack. |
| Compare conformations in a stack. |
| Recaluclate energies in a stack. |
| Impose a conformation from a stack onto an object. |
| Generate stack of normal mode backbone movements. |
© 2026 All Rights Reserved MolSoft LLC Terms of Use | Privacy Policy



