[ Object | only | parray | pattern | png | Pdb | peptide bond | pharmacophore | profile | prosite | rarray | Psa | REBEL | real | regularization | residue | rgb | ribbon ]
A pdb file or a chemical molecule with 3D coordinates, or a newly build group of molecules that can co-exist in space are called objects in ICM. Please do not confuse this specific use of the word with general shell objects that include other data types. A session may contain any number of molecular objects.Each object (i.e. molecular object) in turn consists of three levels or organization:
- molecules
- residues
- atoms
ICM understands expressions specifying different selections in objects, see object selections. There are two main classes of objects:
- ICM objects
- non-ICM objects
show energy , minimize and montecarlo .
The non-ICM objects (from the second line on) may have subtypes such as
"ICM" ready for energy calculations. Those objects are either built in ICM or converted to the ICM-type.
|
"X-Ray" determined by X-ray diffraction
|
"NMR" determined by NMR
|
"Model" theoretical model (watch out!)
|
"Electron" determined by electron diffraction
|
"Fiber" determined by fiber diffraction
|
"Fluorescence" determined by fluorescence transfer
|
"Neutron" determined by neutron diffraction
|
"Ca-trace" upon reading a pdb, ICM determines if an object is just a Ca-trace.
|
"Simplified" special object type for protein folding games.
|
Each object contains the following information:
- Molecules (with their own internal structure, including residues and atoms)
- name ( changed by
renameand returned by theName( os ) function ) - remark (set by
set commentand returned by theNamex( os ) function ) - header ( returned by the
Header(~~os ) function ) - filename ( returned by the
File( os ) function ) - date ( return by the
Datefunction ) - resolution ( returned by the
Resolutionfunction ) - type (returned by the
Type( os 2) function )
Functions on molecular objects. os_ means object selection for other abbreviations see the link.(note that some functions that also operate molecules, residues and atoms are not included).
`Cell ( { os | m_map } )
`Charge ( { os | ms | rs | as } [ formal | mmff ] )
`Date ( os )
`Field( { rs | ms | os } [ i_fieldNumber ] )
`Field( { as | rs | ms | os } [ s_fieldName ] )
`Field ( os i_fieldNumber )
`File ( os )
`Label ( os_objects )
`Mass( as | rs | ms | os )
`MaxHKL( { map | os | [ R_6CellParameters ] }, r_minResolution ) → I3
`Mol ( { os | rs | as } )
`Name ( s_hint os unique )
`Name ( os )
`Name ( s_root os_1 unique )
`Namex ( os )
`Next ( { as | rs | ms | os } )
`Nof( { os | ms | rs | as | vs } )
`Nof( { os | ms | rs } [ atom ] )
`Parray ( object [os] )
`Res ( { os | ms | rs | as } [ append ] )
`Resolution ( os_object ) → R_array_of_resolutions
`Select ( as_sourceSelection os_targetObject )
`Select ( os_sourceObject S_residueSelStrings )
`Select ( os_sourceObject I_atomNumbers )
`String( { os | ms | rs | as } [ i_number ] )
`Symgroup ( { s_groupName | os_object | m_map } )
`Symgroup ( { i_groupNumber | os_object | m_map } string )
`Transform( s_group|iGroup|os_1|map ) → R_12N_all_fract_transformations
`Transform( s_group|iGroup|os_1|map iTrans ) → R_12_fract_transformation_i
`Type ( os_object , 2 )
`dsCellBox os_Commands acting on objects
append stack os_ligandObject
clear graphic [ os_ ]
convert [ exact ] [ charge ] [graphic] [ tether ] [ os_non-ICM-object ] [ s_newObjectName ]
copy [ strip ] [ tether ] os [ s_newObjectName ] [ delete ]
delete { object | os }
find pdb rs_fragment os_objectWhereToSearch s_3D_align_mask [ s_sequencePattern [ s_SecStructPattern ] ] [ r_RMSD_tolerance]
find pattern [ number ] [ mute ] s_sequencePattern [ i_mnHits] [ { os_objectWhereToSearch | seq_Name | s_seqNamePattern } ... ]
find segment ms_source [ os_objectWhereToSearch_2] [ r_maxRMSD ] [ i_minNofAlignedResidues [ i_accuracyParam ] ]
move ms_MoleculesToMove os_destination
move os_ObjectToMove os_destination
rename { os [ full ] | ms | rs | as } s_newName
rotate [ os | ms | g_grob ] M_rotation
set comment [ append ] os_Object s_comment # returned by the `Label() function
set object [ os_newObjName ]
set symmetry os_object R_6cell s_symgroup | i_symgroup [ i_NofChains ] # set symmetry os s_crysym_record
set type os s_type
set [ atom ] type mmff [ os]
show { os | ms | rs | as | vs }
sort object R|I # reorder objects by arrays, e.g. --{sort object Resolution(a_*.)}
sort object os_ i_pos # move selected OBJECTS to specified position
sort os [ field = i_Field ] # sort MOLECULES in selected objs by field
strip os_object [ virtual ]
superimpose os_static I_atomNumbers1 os_movable I_atomNumbers2
translate { os | ms | g_grob } [ add ] [ symmetry ] R_3translationVector Important macros working on molecular objects.
convertObject ms l_delete_water l_optimize_hydrogens l_replace_the_original l_display
convert2Dto3D os l_build_hydrogens (yes) l_fixOmegas (yes) l_display (no) l_overwrite (yes)
icmPmfProfile os ( a_ ) l_accessibilityCorrection (yes) l_display ( no )
makeSimpleModel seq ali os
makeSimpleDockObj [ os_object ] [ s_newObjName ]
only commands usually add or append
to the current settings.
Examples:
display only g_icos # undisplay everything which is in the
# graphics window (if any)
# and display icosahedron[ Object parray | Sequence parray | Image parray ]
pointer array, abbreviated as P. An array of pointers to objects. Currently there are several types of parrays, that consist of the following data types:chemicalimagesequence- distances (e.g. interatomic distances, hbonds, angles, torsions)
3d labelsobjectslide
mol or sdf file.
These compounds do not exist outside this array
as independent molecular objects, but can be converted to molecular objects.
creating mol-arrays To create a molarray use the read table mol command and first column of this table will be such an array. Example:
read table mol "Maybridge.sdf"Chemical function.
ICM molecular objects (both converted and unconverted) can be grouped into arrays and shown in tables.
See the following functions:
ICM sequences may be grouped into arrays and placed into tables. Some functions supporting sequence arrays:
Sequence arrayParray( object ... )TostringLengthName sequenceset name sequenceSstructureset sstructure sequenceNamex sequence
ICM provides multiple ways of generating images and performing advanced operations with them: See
Image- Info(image)
make imageset background imageset textureSum imageColor imageName imageNamex imageimage
* # anything until the next element, e.g. Find( "*[!A]?SI*" sequence )
? # any symbol
[XY..] # any one of the listed one-letter codes, e.g. [DE] for asp or glu.
! # negation, e.g. [!PG] anything except P or G
\{l\}, or \{l1,l2\} # anything of length l, or from length l1 to l2, e.g. \{0,5\}
#
# Example:
#
Find( "*A[LV]\{0,5\}*[!DE]?\{8\}C*" sequence )See also:
- find pattern - find a pattern in a single sequence,
- find database pattern - efficient parallel pattern search in a BLAST-formatted sequence databank.
-
Pattern( s_consensus) - create a regular pattern expression from a consensus, -
Pattern( alignment) - create a regular pattern expression from an alignment, - regular expressions and pattern matching
-
prosite- a collection of sequence patterns -
Find( s_pattern sequence ) to find pattern in all loaded sequences
[ pattern-matching ]
graphics image format. Stands for Portable Network Graphics and was designed to replace the GIF format and, to some extent, the much more complex TIFF format. While GIF allows for only 256 palette colors, PNG can handle a variety of color schemes like TIF (1,3,8, 24, etc. bit colors). Furthermore, PNG is free, while GIF is subject to licensing fees. PNG also supports alpha-channel. Since 1998 most browsers correctly display PNG images.
To write a png with transparent background use
write png transparent
Pattern matching and regular expressions. Use the following metacharacters to construct regular expressions (try guess what string is used in the examples!)
- * matches any string including an empty string (e.g. "*see*" )
- ? matches any single character (e.g. "???ee M")
- [string] matches any one of the enclosed characters. Two characters separated by dash represent a range of characters. Examples: [A-Z], [a-Z], [a-z], [0-9] (e.g. "[A-Z] see [A-Z]"
- [ !string] negation. matches any but the enclosed characters (e.g. "I see [!K]")
- single-character multiplication: character\{m,n\} (e.g. "I?\{3,6\}M" - repeat any character, ?, from 3 to 6 times)
a_*.//c?,n,c ), and in
list, group, delete commands.
Note that for the latter three commands the pattern must be quoted.
Bernstein et al., 1977; Abola et al., 1987).
The new citations can be found at http://www.rcsb.org/pdb/ .
On November 20th, 2001 it contained 16596 entries.
On August 21st, 2007 if contained 45368 Structures.
An example ATOM record:
ATOM 52 N HIS D 18 53.555 24.250 49.573 1.00 32.59icm.cnt file.
Pharmacophores in ICM are special objects of the non-ICM type. A pharmacophore can combine special ph4 centers and disconnected chemical fragments.
The ph4 centers have the following atom types and names:
- code:
- 315 Qa HBA
- 316 Qd HBD
- 317 Qm aromatic
- 318 Qh hydrophobic
- 319 Qp positive
- 320 Qn negative
- 321 Qx ExVolume
- 322 Qv a point defining the direction vector from Qa,Qd and Qm, or a pseudo atom
Creating a pharmacophoreA pharmacophore can be created from an object with the makePharma macro,
macro makePharma ms s_name ("pharm") l_points (yes) l_display
The l_points logical defines if the pharmacophoric centers or fragments are created from the source molecule.
The new object gets type pharmacophore (see the
set type os "pharmacophore" Note the following issues:
- for polar atoms with hydrogens and lone pairs, both Qa and Qv centers are created at the s
- the directions of Qa-Qv or Qd-Qv vectors need adjustment
- the direction atoms (Qv) can be selected and removed. In this case the direction will not be matched.
- two or more ph4 objects can be merged into a single pharmacophore object. For example one can merge some ph4-centers with some fragments.
- the properties of points can be edited interactively or with the set as_ commands.
setbfactoras1_ r_radius # to change the tolerance radiusset bfactor a_pharma.p5/5/Qv10 0.8- resetting the radius (stored in the
bfactorfield) for the direction pointQvwill change the width of the direction tolerance. - you can also change point type and name, e.g.
set type a_pharma.//Qa3 316 rename a_pharma.//Qa3 "Qd4" - the coordinates can be reset with the
set atomcommand.
The following commands and functions work with pharmacophores:
find pharmacophore:find pharmacophore as_pharmQuery chemarray3Dsuperimpose:superimpose os_ph4obj ms_ligand pharmacophoreChemical:
# creates pharmacophore, this function is used inChemical( ms|os pharmacophore )makePharmaRmsd:Rmsd ( as_pharmTemplate as_select2 pharmacophore )makePharmamacro- makePharmaAtomsmacro # generates separate pharma centers from selections
Saving ph4 objects.
The objects can be saved with the regular write object command or write binary commands.
Abagyan, Frishman, and Argos, 1994).
One can add two profiles ( prf1 + prf2 ), multiply them (prf1 * prf2),
concatenate two profiles (prf1//prf2), and extract a
part of a profile ( prf[15:67] ).
Profile can be read from a .prf file and calculated from an alignment with the
Profile() function.
To make a table out of a profile use this example:
make sequence 10; align sequence; prf=Profile(aln);
t = Table(Matrix(prf)) # columns names 'A','B','C',...
add column t Count(Nof(t)) Split(String(Sequence(prf)),"") name={"ipos","seq"} # adds number and cons seqSequence()
Consensus()
Matrix()
Align().
"C?\{4,5\}CCS??G?CG????[FYW]C".
Included in ICM is only the old release 13 (1995). Until the 15th release the files were fully open for redistribution and said this.
CC *************************************************************************
CC
CC This file may be copied and redistributed freely, without advance
CC permission. You are allowed to reformat it for use with a software
CC package, but you should not modify its content without permission
CC from the author).
CC
CC *************************************************************************If you want to use a newer version of prosite and your organization is non-profit or has a license from SIB, you can download prosite.dat from, e.g.:
unix wget ftp://us.expasy.org/databases/prosite/release_with_updates/prosite.dat
read prosite "./prosite.dat" # read the newer version in
readUniprot "IL2_HUMAN"
find prosite IL2_HUMANSee also:
- read prosite,
-
s_prositeDat, - find prosite - find all prosite patterns in a single sequence,
- find profile - find all prosite profiles in a single sequence
ICM class for arrays of real numbers: { -1.6 , 2.150 3., -160.}
An array can be formed dynamically from simple reals with the double slash (concatenate) operator, e.g. 1.3//-1.//dielConst//14.
Real arrays also support special values (see rarray constant )
ND(not defined)- >r_value
- <r_value
INF: infinity- -INF : infinity
Example:
add column t {1.}//ND//Real(">2.")//Real("<3.")//Real("INF")//Real("-INF")//2.3
add column t
See also: Rarray , Real , Toreal , read rarray , rarray constant , GUI.displayNDValueStyle
Polar surface area or PSA is a parameter important for predicting transport properties of drugs. It correlates with intestinal absorption and blood brain barrier penetration. (e.g. see Polar Surface Area by Peter Ertl in Molecular drug properties: measurement and prediction, Raimund Mannhold, Ed, Wiley-VCH, v 37, 2008 )
Single conformation 3D PSAPSA is calculated as the exposed van der Waals surface of oxygens and nitrogens as well as the hydrogen atoms attached to them. For a converted and current ICM object PSA can be calculated with the following command:
show surface area a_//n*,o* | ( a_//h* & Next( a_//n*,o* bond )) a_ mute vwExpand = 0.
PSA = r_out # the previous command returns the value in r_outThe conversion is needed only to make sure that all hydrogens are in place. If they are, the conversion is not really needed and the same command can be applied.
Multiple conformation 3D PSAThis measure obviously depends on the conformation of the molecule. An average PSA can be calculated with the following script from a stack on conformers generated by a montecarlo command, e.g.:
for iconf=1,Nof(conf)
load conf iconf
show surface area a_//n*,o* | ( a_//h* & Next( a_//n*,o* bond )) a_ mute vwExpand = 0.
avPSA += r_out
endfor
avPSA /= Nof(conf)_confGen script.
Fast and accurate estimate of PSA from chemical topology only ( TPSA or MolPSA )
Ertl et al. (see the reference above) proposed a way to estimate the PSA value directly from a two dimensional
topology of a molecule without a need to convert it to 3D and sample its geometrical states.
The ICM MolPSA function uses a similar method to estimate PSA, ( e.g. Predict( Chemical("CCO"),"MolPSA" ) ).
The following method was used to derive a model (see the learn command ):
- Six thousand drug compounds were converted to 3D, optimized and their real single conformation PSA values were evaluated
- The chemical topologies were converted into Molsoft fingerprints and trained against the computed PSA values
- The resulting model had a cross validated squared correlation coefficient of predicted vs training values of Q2=0.99 and the root mean square error of Rmse=1.56.
read table mol s_icmhome+"/moledit/Dictionary.sdf" name="t"
add column t Predict(t.mol "MolPSA" ) name="MolPSA"
show Mean(t.MolPSA)
a method to solve the Poisson equation for a molecule. REBEL is a new powerful implementation
of the boundary element method with analytical molecular surface as dielectric
boundary. This method is fast (takes seconds for a protein) and accurate.
REBEL stands for Rapid Exact-Boundary ELectrostatics.
The energy calculated by this method consists of the Coulomb energy and the
solvation energy which is returned in the
r_out system variable.
|
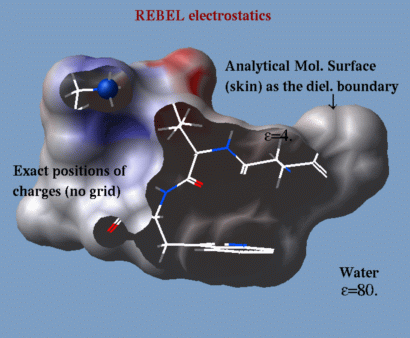
|
-
electroMethod= "boundary element"; - .`TOOLS.rebelPatchSize (1., but can be smaller, say 0.3 or 0.5, it is a tradeoff between speed and accuracy)
-
dielConst(the default is usually OK); -
dielConstExtern(the default is usually OK); - set charge as_ r_Charge (modify charges if you like);
- make boundary (if you want to make
several evaluations of energy or
Potential( ) with the same boundary. The "boundary" parameters depend only on conformation and do NOT depend on charges. You can redefine charges afterwards and get a corrects energy evaluation); - delete boundary (if you do not need it);
- show energy (make sure the "el" term is on);
-
Potential( as_targets as_charges) (if the boundary exists, returns potentials from charges at the target atoms); - color grob potential [heavy] (create graphics
object, say, with
make grob skin
actually it can be anygroband color it by the REBEL potential);optionheavyresults in better coloring. - Matrix(boundary) : returns the matrix solved to find the boundary element charges.
dsRebel: a macro for calculating the potential-colored electrostatic surface.
The polarization charges, or induced surface charges, can be returned by the Rarray( as_ )
function after the equation is solved, e.g.:
electroMethod = 4
show energy "el"
Rarray( a_//* ) # returns polarization chargesElectrostatic troubleshooting
Problem: the electrostatic surface looks different before and after conversion to ICM .
Explanation: The unconverted PDB object has (i) an incomplete set of atoms ( lacking hydrogens for the X-ray entries, occasional missing side chains or loops, etc.); (ii) has formal charge ambiguities for his (hip), asp (ash) etc. , see below (iii) has no charges, i.e. the formal and partial charges are not assigned;
The dsRebel procedure
(i) builds a full atomic model with hydrogens,
(ii) resolves at least some residue charge ambiguities, and
(iii) assigns to the atoms of the unconverted objects a simplified set of formal charges.
See convertObject for further details.
The difference between the formal charges of the unconverted and the full distributed partial charges after conversion creates a small difference in electrostatic pattern.
If the conversion procedure charges a residue (e.g. a cysteine next to a metal ion), then the electrostatic patterns will differ much more in the vicinity of that residue.
Problem: the electrostatic surface looks different for different PDBs of the same protein
Explanation 1: Some residues can be charged or uncharged. In a specific PDB can be marked by a different 3-letter residue name or not. For example:
| Amino Acid | Frequent | Rare |
|---|---|---|
| aspartate | asp (-) | ash (0) |
| cysteine | cys (0) | cym (-) |
| glutamate | glu (-) | glh (0) |
| histidine | his (0) | hip (+) |
| lysine | lys (+) | lyn (0) |
| tyrosine | tyr (0) | tym (-) |
hip while in another they are simply marked as his . In this case the total charge will change and the dsRebel pictures will be different.
Problem: the numerical singularities are reported
Sometimes numerical instabilities inherent to the REBEL calculation lead to singularities and strange values
of electrostatic potential. This can be detected by looking at the polarization charge values.
The ePotProtected macro in the _rebel file contains the code that checks for this type of instabilities.
12.3, 2.0, -4.501 ). There are a number of
predefined real variables.
Reals can be concatenated into a real array like this: 12.3//2.0//-4.5
Reals may be mentioned in
arithmetic expressions,
commands and
functions.
Examples:
a = -1.2
b = Abs(Sin( 2.3 * a - 3.0 / a))icm.res residue library)
to the atom positions of a target PDB structure (usually provided
by X-ray crystallography or NMR). Regularization is required because
the experimentally determined PDB-structures often lack hydrogen atoms
and positional errors may result in the unrealistic van der Waals
energy even if these structures were energetically refined (since
the refinement of the crystallographic structures typically ignores
hydrogen atoms and employs different force fields). The following
steps are required to create the regularized and energy refined
ICM-model of an experimental structure:
- an extended all-atom model of a particular protein is generated
with regular geometry characteristics (see the
buildcommand and theIcmSequencefunction); - the non-hydrogen atoms in the model are assigned to the equivalent atoms in the model (see set tether);
- the regularized structure is built starting from the N-terminus by adding atoms one-by-one (see minimize tether);
- methyl groups are rotated to reduce van-der-Waals clashes;
- combined geometry and energy function is optimized;
- polar hydrogen positions are adjusted;
- optionally the model may be additionally minimized, now without tethers to observe a "stability" of the model in the local energy minimum.
regul .
icm.res file.
You may create your own residues with the write library command.
Residues can be selected with the ICM-selection expression
(e.g. a_/ala, a_/15, a_/15:20, a_/"RDGE" etc.),
labeled with the display residue label rs_ command,
by double clicking with the right mouse button, via a pop-up menu, or from
* Added tothe registry{ Table( model chem [inverse] ) } function.
the GUI menu.
tif,
targa,
postscript.
|
a graphical representation of a polypeptide chain backbone by a smooth
solid ribbon. DNA and RNA can be also displayed in a ribbon style.
There are three types of elements of the ribbon display depending on the secondary structure assigned to a given residue. |

|
Residues marked as alpha-helices ('H') will be shown by a flat ribbon, those marked as beta-sheets ('E') will be flat ribbon with an arrow-head, and the rest will be shown by a cylindrical "worm". The ICM-shell parameter
GRAPHICS.wormRadius defines its radius.
Default ribbon colors are defined in the icm.clr file.
Note that minor secondary structure elements like 3/10 helix ('G'),
Pi-helix ('I') are colored by the corresponding colors
( the threetenRibbn and piRibbon parameters in the icm.clr file), 'Y' type
is colored by the alphaRibbon color, and 'L','P' and
'B' (isolated beta-residue) residues are colored by the "betaRibbon" color.
DNA and RNA ribbons are colored according to the base type:
A-red, C-cyan, G-blue, T or U - gold.
Preference ribbonStyle allows one to display a simplified
segment representation of the secondary structure elements instead of
(or together with) the ribbon.
If the polypeptide chain has a break (missing residues) the ribbon
shows bullets or nothing depending on the GRAPHICS.chainBreakStyle .
The DNA/RNA ribbons consists of two parts the backbone ribbon
and the bases shown with the sticks and balls. To selectively
display and undisplay the bases, you can do the following:
Example:
read pdb "1dnk" # contains 2 dna mol.
display ribbon a_1.2,3 # both bases and backbone
undisplay ribbon base a_1.2 # bases disappear
display ribbon base only a_1.2 # only bases
display ribbon a_1.2,3 yellow # both bases and backbone
color ribbon a_1.3 magenta # the second chain backbone
color ribbon a_1.2,3 bases # default by base type
cool a_ # cool is a rich macro. View the whole thing-
display ribboncommand (alsoundisplayandcolor) -
GRAPHICS.ribbonRatioparameter -
GRAPHICS.ribbonWidthparameter -
GRAPHICS.ribbonCylinderRadiusparameter -
GRAPHICS.dnaRibbonRatioparameter -
GRAPHICS.dnaRibbonWidthparameter -
GRAPHICS.ribbonGapDistanceparameter -
GRAPHICS.dnaRibbonWormlogical -
GRAPHICS.ribbonWormlogical -
l_breakRibbonlogical -
GRAPHICS.chainBreakLabelDisplaypreference -
GRAPHICS.chainBreakStylepreference -
GRAPHICS.dnaRibbonStylepreference -
ribbonColorStylepreference -
ribbonStylepreference - residue field "_WORMRADIUS_" for variable thickness