[ Abs | Acc | Acos | Acosh | Align | Angle | Area | Asin | Asinh | Ask | Askg | Atan | Atan2 | Atanh | Atom | Augment | Axis | Blob | Bfactor | Boltzmann | Box | Bracket | Cad | Ceil | Cell | Charge | Chemical | Cluster | Color | Compare | Consensus | Contour | Corr | Cos | Cosh | Count | CubicRoot | Date | Deletion | Descriptor numeric | Descriptor | Det | Disgeo | Distance | Eigen | Energy | Entropy | Error | Error soap | Exist | Existenv | Extension | Exp | Field | File | Find | Floor | Formula | Getarg | Getenv | Gradient | Grob | Group | Header | Histogram | Iarray | IcmSequence | Image | InChi | Index | Indexx | Insertion | Info | Info image | Info model | Integer | Integral | Interrupt | Label | Laplacian | Length | GaussFit | LinearFit | LinearModel | Log | Map | Mass | Moment | Match | Matrix | Max | MaxHKL | Median | Mean | Min | Money | Mod | Mol | Name | Namex | Next | Nof | Norm | Normalize | NotInList | Obj | Occupancy | Path | Parray | Pattern | Pi | Potential | Power | Predict | Probability | Profile | Property | Putarg | Putenv | Radius | Random | Rarray | Real | Remainder | Reference | Replace | Res | Resali | Resolution | Ring | Rfactor | Rfree | Rmsd | Rot | Sarray | Score | Select | Sequence | Shuffle | Sign | Sin | Sinh | Site | Slide | Smiles | Smooth | SolveQuadratic | SolveQubic | Sql | Sqrt | Sphere | SoapMessage | Sort | Split | Srmsd | String | Sstructure | Sum | Symgroup | Table | Tan | Tanh | Tensor | Temperature | Time | Tointeger | Tolower | Toreal | Torsion | Tostring | Toupper | Tr123 | Tr321 | Trace | Trans | Transform | Transpose | Trim | Trim chemical | Trim sequence | Turn | Type | Unique | Unix | Value | Value soap | Vector | Version | Volume | View | Warning | Xyz ]
ICM-shell functions are an important part of the ICM-shell environment. They can be divided into hardwired built-in functions and open-shell functions written as an icm script ( seeicm shell functions ).
They have the following general format:
FunctionName ( arg1, arg2, ... ) integer, real, string, logical, iarray,
rarray, sarray, matrix, sequence, profile, alignments,
maps, graphics objects, a.k.a. grob and selections.
The order of the function arguments is fixed in contrast to that of commands. The same function may perform different operations and return ICM-shell constants of different type depending on the arguments types and order. ICM-shell objects returned by functions have no names, they may be parts of algebraic expressions and should be formally considered as 'constants'. Individual 'constants' or expressions can be assigned to a named variable. Function names always start with a capital letter. Example:
show Mean(Random(1.,3.,10)) Abs ( real ) real absolute value.
Abs ( integer ) integer absolute value.
Abs ( rarray ) rarray of absolute values.
Abs ( iarray ) iarray of absolute values.
Abs ( map ) map of absolute values of the source map.
Examples:
a=Abs(-5.) # a=5.
print Abs({-2.,0.1,-3.}) # prints rarray {2., 0.1, 3.}
if (Abs({-3, 1})=={3 1}) print "ok"Acc function just uses surface values, it does not reevaluate them.
Therefore, make sure that the show area command (or show energy,
minimize , etc. with the "sf" surface term turned on),
has been executed before you use the Acc function.
If you specify the threshold explicitly, it must range from 0.0 to 1.0, otherwise it is set to 0.25
for residue selections and 0.1 for atom selections.
Acc ( rs , [ r_Threshold ] ) - returns
residue selection, containing a subset of specified residues `rs_
for which the ratio of their current accessible surface to the standard exposed surface is greater than
the specified or default threshold (0.25 by default).
ICM stores the table of standard residue accessibilities in an unfolded state calculated
in the extended Gly-X-Gly dipeptide for all amino acid residue types.
It can be displayed by the show residue type command,
or by calling function Area( s_residueName ),
and the numbers may be modified in the icm.res file.
The actual solvent accessible surface, calculated by a fast dot-surface algorithm, is divided by the standard one and the residue gets selected if it is greater than the specified or default threshold. ( r_Threshold parameter is 0.25 by default).
Acc ( as_select, [ r_Threshold ] ) - returns
atom selection,
containing atoms with accessible surface divided by the total surface of the
atomic sphere in a standard covalent environment greater than the
specified or default threshold (0.1). Accessibility at this level does not make
as much sense as at the residue level. The standard surface of the atom
was determined for standard amino-acid residues. Note that hydrogens were NOT
considered in this calculation. Therefore, to assign surface areas to the
atoms use
show surface area a_//!h* a_//!h* command or the
show energy "sf" command.
You may later propagate the accessible atomic layer by applying
Sphere( as_ , 1.1), where 1.1 is larger than a typical
X-H distance but smaller than the distance between two heavy atoms.
(the optimal r_Threshold at the atomic level used as the default is 0.1,
note that it is different from the previous ).
Examples:
# let us select interface residues
read object s_icmhome+"complex"
# display all surface residues
show surface area
display Acc( a_/* )
# now let us show the interface residues
display a_1,2
color a_1 yellow
color a_2 blue
show surface area a_1 a_1 # calculate surface of
# the first molecule only
# select interface residues
# of the first molecule
color red Sphere(a_2/* a_1/* 4.) & Acc(a_1/*)
read object s_icmhome+"crn"
show energy "sf"
display
display cpk Acc(a_//* 0.1) # display accessible atoms
show surface area # prior to invoking Acc function
# surface area should be calculated
color Acc(a_/*) red # color residues with relative
# accessibility > 25% red Acos ( real | integer ) real arccosine of its real or integer argument.
Acos ( rarray ) rarray of arccosines of rarray elements.
Examples:
print Acos(1.) # equal to 0.
print Acos(1) # the same
print Acos({-1., 0., 1.}) # returns {180. 90. 0.} Acosh ( real | integer ) real inverse hyperbolic cosine of its real or integer argument.
Acosh ( rarray ) rarray of inverse hyperbolic cosines of rarray elements.
Examples:
print Acosh(1.) # returns 0
print Acosh(1) # the same
print Acosh({1., 10., 100.}) # returns {0., 2.993223, 5.298292}[ sequence | structural alignment | sub_alignments ]
family of the alignment functions. These function return analignment icm-shell object and perform
- sequence alignment (with the
Needleman and Wunschalgorithm with zero gap end penalties ( ZEGA ), - structural alignment, or
- sub-alignment extraction
- : Align(
[ | area ] ) => - : Align(
{ distance | superimpose | static [ (18) (0.5)] } ) => - : Align( [selection] ) =>
; first two (selected) sequences - : Align(
) => ; sub-alignment of seq1 vs seq2 - : Align(
) => - : Align(
dash|compress ) => - : Align(
) => # single sequence alignment - : Align(
Align ( [ sequence1, sequence2 ] [ area ] [ M_scores ] )
- returns ZEGA- alignment.
If no arguments are given, the function aligns the first two sequences in the sequence list.
For sequence alignments, the ZEGA-statistics of
structural significance ( Abagyan, Batalov, 1997) is given and can be additionally
evaluated with the Probability function.
The reported pP value is -Log(Probability,10).
Returned variables:
i_out- the number of identical residues in the alignmentr_out- containsLog(Probability_of_structural_dissimilarity ) only for pairwise alignmentsr_2out- percent identity of the alignment.
Simple pairwise sequence alignment
Align( ) Align( seq1 seq2 ) alignMethod preference allows you to perform two types of pairwise sequence alignments: "ZEGA" and "H-align". If you skip the arguments, the first two sequence are aligned.
Example:
read sequences s_icmhome+"sh3.seq" # read 3 sequences
print Align(Fyn,Spec) # align two of them
Align( ) # the first two
a=Align( sequence[1] sequence[3] ) # 1st and 3rd
if(r_out > 5.) print "Sequences are struct. related"
Aligning DNA or RNA sequencesMake sure to read the dna.comp comp_matrix before using the Align function, e.g.
a=Sequence("GAGTGAGGG GAGCAGTTGG CTGAAGATGG TCCCCGCCGA GGGACCGGTG GGCGACGGCG")
b=Sequence("GCATGCGGA GTGAGGGGAG CAGTTGGGAA CAGATGGTCC CCGCCGAGGG ACCGGTGGG")
read comp_matrix s_icmhome+"dna.cmp"
c = Align(a,b)Aligning with custom residue weights or weights according to surface accessible area
Align( seq1 seq2 area ) Option
area will use relative residue accessibilities to weight the residue-residue
substitution values in the course of the alignment (see also accFunction ).
The weights must be positive and less than 2.37 . Try to be around or less than 1. since relative accessibilities are always in [0.,1.] range. Values larger than
2.37 do not work well anyway with the existing alignment matrices
and gap parameters.
Use the Trim function to adjust the values, e.g. Trim( myweights , 0.1,2.3 ) ).
E.g.
read pdb "1lbd"
show surface area
make sequence
Info> sequence 1lbd_m extracted
1lbd_a # see the relative areas
read pdb sequence "1fm6.a/" # does not have areas
Info> 1 sequence 1fm6_a read from /data/pdb/fm/pdb1fm6.ent.Z
ali3d = Align( 1lbd_a 1fm6_a area )set area seq1 R_weights # must be > 0. and less than 2.37
Align( seq1 seq2 area )Introducing positional restraints into the alignment matrix
Align( seq1 seq2 M_positionalScores ) If sequence similarity is in the "twilight zone" and the alignment is not obvious, the regular comp_matrix{residue substitution matrix} is not sufficient to produce a correct alignment and additional help is needed. This help may come in a form of the positional information, e.g. histidine 55 in the first sequence must align with histidine 36 in the second sequence, or the predicted alpha-helix in the first sequence preferably aligns with alpha-helix in the second one.
In this case you can prepare a matrix of extra scores for each pair of positions in two sequences, e.g.
seq1 = Sequence("WEARSLTTGETGYIPSA")
seq2 = Sequence("WKVEVNDRQGFVPAAY")
Align()
# Consensus W.#. .~~.~G%#P^
WEARSLTTGETGYIPS--
WKVE--VNDRQGFVPAAY
m = Matrix(17,16,0.)
m[10,4] = 3. # reward alignment of E in seq1[10] and E in seq2[4]
Align(seq1 seq2 m )
# Consensus W.# E ~G%#P^
WEARSLTTGE----TGYIPS--
WKV------EVNDRQGFVPAAY The
AlignSS shell function shows a more elaborate example in which extra scores are prepared
to encourage alignments of the same secondary structure elements.
Warning. The alignment procedure is rather subtle and may be sensitive to the gap parameters and the comparison matrix. Avoid matrix values comparable with gap opening penalty.
See also: Probability( ali .. ) for local alignment reliability.
Two types of structural alignments or mixed sequence/structural alignments
can be performed with the Align function.
Align( seq_1 seq_2 distance [ i_window ] [ r_seq_weight ] ) Align( seq_1 seq_2 superimpose [ i_window ] [ r_seq_weight ] ) In both cases the function uses the dynamic algorithm to find the alignment of the locally structurally similar backbone conformations.
The alignment based on optimal structural superposition of two 3D structures may be different from purely sequence alignment
Preconditions:
- sequences must be linked to 3D molecules to access the coordinate information;
- two 3D structures must have superposable subsets
See also: align ms1 ms2 function
Deriving an alignment from tethers between two 3D objects
Align ( ms ) alignment between sequences of the specified molecule and the template molecule
to which it is tethered. The alignment is deduced from the tethers imposed.
Example:
build string "se ala his leu gly trp ala" name="a" # obj. a
build string "se his val gly trp gly ala" name="b" # obj. b
set tether a_2./1:3 a_1./2:4 align # impose tethers
show Align(a_2.1) # derive alignment from tethers
write Align(a_2.1) "aa" # save it to a file
Align ( ali, seq_1, seq_2 ) alignment of the input alignment ali_,
reorders of sequences in the alignment according to the order of arguments.
Extracting a multiple alignment of a subset of sequences from a multiple alignment
Align ( ali, I_seqNumbers ) alignment .
Sequences are taken in the order specified in I_seqNumbers.
Examples:
# 14 sequences
read alignment msf s_icmhome + "azurins"
# extract a pairwise alignment by names
aa = Align(azurins,Azu2_Metj,Azur_Alcde)
# reordered sub-alignment extracted by numbers
bb = Align(azurins,{2 5 3 4 10 11 12})Resorting alignment in the order of sequence input with the
Align ( ali_, I_seqNumbers ) function. Load the following macro and apply it to your alignment. Example:
macro reorderAlignmentSeq( ali_ )
nn=Name(ali_) # names in the alignment order
ii=Iarray(Nof(nn))
j=0
for i=1,Nof(sequence) # the original order
ipos = Index( nn, Name(sequence[i] ) )
if ipos >0 then
j=j+1
ii[j] = ipos
endif
endfor
ali_new = Align( ali_ ii )
keep ali_new
endmacro Angle ( as_atom ) print Angle( # and then click the atom of interest. Angle ( as_atom1 , as_atom2 , as_atom3 ) Angle ( R_3point1 , R_3point2 , R_3point3 ) Angle ( R_vector1 , R_vector2 )
Angle ( as table ) read pdb "1xbb"
t=Angle(a_H table)
sort t.angle
show t
Angle ( as|rs|ms|os as_filter error ) rarray of minimal angles within each specific unit of the selection.
The size of the array depends on the level of the selection. Used to detect errors (too small angles).
Examples:
d=Angle( a_/4/c ) # d equals N-Ca-C angle
print Angle( a_/4/ca a_/5/ca a_/6/ca ) # virtual Ca-Ca-Ca planar angle
The rotation angle corresponding to a transformation vector is returned as r_out by
the Axis( R_12 ) function.
Area( grob [error] ) → r
Area( as | rs ) → R_atomAreas|R_resAreas # needs surface calculation beforehand
Area( rs type ) → R_maxAreas_in_GLY_X_GLY
Area( as R_typeEyPerArea energy ) → R_atomEnergies
Area( seq ) → R_relAreasPerResidue
Area( s_icmResType ) → r
Area( rs rs_2 ) → M_contactAreas
Area( rs rs_2 distance [ min(4.) max(8.) [Ca_Cb_len(2.3)]] ) → M_0_to_1_contact_strength
Note that if an atom selection is provided as an argument the surface area needs to be computed beforehand with
the show area or show energy "sf" command. The detailed description can be found below:
Area ( grob [error] ) real surface area of a solid graphics object.
Option error makes it return the fraction of the surface that is not closed to detect the holes or missing
patches in what supposed to be a closed surface. (e.g.
g = Grob("SPHERE",1.,2)
show Area(g)
if(Area(g error)>0.01) print "Surface not closed" # check for holesSee also: the
Volume( grob ) function, the split command and
How to display and characterize protein cavities
section.
Area ( as [ [ R_userSolvationDensities ] [ energy ] ] ) rarray of pre-calculated solvent accessible areas or energies for selected atoms `as_ .
This areas are set by the show area surface|skin of show energy "sf" commands.
Make sure to clean up the areas with the set area a_//* 0. command before computing the areas
with show energy command since the command ignores hydrogens.
With option energy returns the product of
the individual atomic accessibilities by the atomic surface energy density. The values of the density
depend on the surfaceMethod preference and are stored in the icm.hdt file. The "contant tension"
value of the preference is a trivial case in which all areas are multiplied by the surfaceTension parameter.
For the "atomic solvation" and "apolar" styles, the densities depend on atom types.
Normally the atomic solvation densities are taken from the icm.hdt file where the density values are listed
for each hydration atom type for "atomic solvation" and "apolar" styles. However, you can provide
your own array of n values R_userSolvationDensities with the number of elements less or equal to the
number of types to overwrite the first n types.
Examples:
read object s_icmhome+"crn.ob"
set area a_//* 0.
surfaceMethod = "apolar"
show energy "sf" # only heavy atoms, alternatively: show surface area mute
Area( a_/15:30/* ) # areas of this atoms
#
# Now let us redefine the first three solvation parameters
# of icm.hdt and calculated E*A contributions of selected atoms
#
Area( a_/15:30/* {10., 20. 30.} energy) Area ( rs ) rarray of pre-calculated solvent accessible areas for selected residues `rs_ .
These accessibilities depend on conformation.
Area ( rs type ) rarray of maximal standard solvent accessible areas for selected residues `rs_ .
These accessibilities are calculated for each residue in standard extended conformation
surrounded by Gly residues. Those accessibilities
depend only on the sequence of the selected residues and do NOT depend on its conformation.
To calculate normalized accessibilities, divide Area( rs_ ) by Area( rs_ type )
Example:
read object s_icmhome+"crn.ob"
show surface area
a=Area(a_/* ) # absolute conformation dependent residue accessibilities
b=Area(a_/* type ) # maximal residue accessibilities in the extended conformation
c = a/b # relative (normalized) accessibilities
Area ( resCode ) → r_standard_area
- returns the real value of solvent accessible area for the specified residue type in the standard
"exposed" conformation surrounded by the Gly residues, e.g. Area("ala").
It is the same value as the Area( .. type ) function.
Area( seq ) → R_relAreasPerResidue
- returns an array of relative areas per residue stored with the sequence by the make sequence command from
molecules in which the areas had been computed beforehand. Note that the sequence keeps only a very limited
accuracy areas. Example:
read pdb "1crn"
show area surface
make sequence # 1crn_a now has relative areas
group table t Sarray( a_/* residue) Area(1crn_a) Area(a_/*)/Area(a_/* type)
show t
Important : "pre-calculated" above means that before invoking
this function, you should calculate the surface by
show area surface
,
show area skin
or
show energy "sf"
commands.
Examples:
build string "se ala his leu gly trp lys ala"
show area surface # calculate surface area
a = Area(a_//o*) # individual accessibilities of oxygens
stdarea = Area("lys") # standard accessibility of lysine
# More curious example
read object s_icmhome+"crn.ob"
show energy "sf" # calculate the surface energy contribution
# (hence, the accessibilities are
# also calculated)
assign sstructure a_/* "_"
# remove current secondary structure assignment
# for tube representation
display ribbon
# calculate smoothed relative accessibilities
# and color tube representation accordingly
color ribbon a_/* Smooth(Area(a_/*)/Area(a_/* type) 5)
# plot residue accessibility profile
plot Count(1 Nof(a_/*)) Smooth(Area(a_/*)/Area(a_/* type) 5) displayAcc( ) function.
(also see the simplified distance-based contacts strength calculation below)
Area ( rs_1 rs_2 ) rarray of areas of contact between selected residues.
You can do it for intra-molecular residue contacts, in which case
both selections should be the same, i.e. Area(a_1/* a_1/*) ;
or, alternatively, you can analyze intermolecular residue contacts,
for example, Area(a_1/A a_2/A).
See also the
Cad function, and example in
plot area in which a contact matrix is
calculated via interatomic Ca-Ca distances.
The table of the pairwise contact area differences is written to
the
s_out string which can later be read into
a proper table via:
read column group name="aa" input=s_out and sorted by the area (see below).
Example:
read object s_icmhome+"crn.ob" # good old crambin
s=String(Sequence(a_/A))
PLOT.rainbowStyle="blue/rainbow/red"
plot area Area(a_/A, a_/A) comment=s//s color={-50.,50.} \
link transparent={0., 2.} ds
read object s_icmhome+"complex"
plot area Area(a_1/A, a_2/A) grid color={-50.,50.} \
link transparent={0., 2.} ds
Area( rs rs_2 distance [ min(4.) max(8.) [Ca_Cb_len(2.3)]] ) → M_0_to_1_contact_strength - evaluates the strength of residue contact based on the projected and extended Ca-Cb vector. It works with both converted and unconverted objects and needs ca, c, and n atoms for its calculation only to be independent on the presense of Gly residues.
By default the procedure finds a point about 1.5 times beyond Cb along the Ca-Cb vector (2.3A) and calculates the distance matrix between those point. Then the distances are converted into the contact strength:
- 0. for distances larger than max_distance (default 8. A)
- 1. for distances smalle than min_distance (default 4. A)
- ( max- dist )/( max - min ) for distances between max and min
read pdb "1crn"
m = Area( a_/A a_/A distance 4. 7. 2.5 )read pdb "1crn"
read pdb "1cbn"
make sequence a_*.A
aln=Align(1crn_a 1cbn_a)
m1=Area( a_1crn.a/!Cg a_1crn.a/!Cg distance ) # !Cg excludes non-matching gapped regions
m2=Area( a_1cbn.a/!Cg a_1cbn.a/!Cg distance )
diff = Sum(Sum(Abs(m1-m2)))/Sum(Sum(Max(m1,m2)))
simi = 1.-diff
printf " Info> dist=%.2f similarity=%.2f or %1f%\n" diff simi,100.*simi
Asin ( real | integer) - returns the
real
arcsine of its real or integer argument.
Asin ( rarray ) - returns the
rarray
of arcsines of rarray elements.
Examples:
print Asin(1.) # equal to 90 degrees
print Asin(1) # the same
print Asin({-1., 0., 1.}) # returns {-90., 0., 90.} Asinh ( real) - returns the
real
inverse hyperbolic sine of its real argument.
Asinh ( rarray) - returns the
rarray of inverse hyperbolic sines of rarray elements.
Examples:
print Asinh(1.) # returns 0.881374
print Asinh(1) # the same
print Asinh({-1., 0., 1.}) # returns {-0.881374, 0., 0.881374} Ask( s_prompt, i_default ) - returns entered
integer or default.
Ask ( s_prompt, r_default ) - returns entered
real or default.
Ask ( s_prompt, l_default ) - returns entered
logical or default.
Ask ( s_prompt, s_default [simple] ) - returns entered
string or default. Option simple suppressed interpretation of the input and makes quotation marks unnecessary by automatically adding quotes around your input text.
Examples:
windowSize=Ask("Enter window size",windowSize)
s_mask=Ask("Enter alignment mask","xxx----xxx")
grobName=Ask("Enter grob name","xxx")
display $grobName
show Ask("Enter string, it will be interpreted by ICM:", "")
#e.g. Consensus( myAlignm )
show Ask("Enter string:", "As Is",simple)
#your input taken directly as a string
See also: Askg
interactive input function that generates a GUI dialog. Return entered text
Askg( s_prompt, i_default ) → s_returnsTheInputStringE.g.
Askg( "Enter your name", "" ) # empty default
Askg( "Enter your name", "Michael" )Return the pressed button.
Askg( s_Question, "Reply1/Reply2/.." simple ) → s_theReply
Makes a GUI dialog with the question and several alternatives
separated by a slash.
This dialog returns one of the string selected
,e.g. "Yes", "No" , or "Cancel" for the "Yes/No/Cancel" argument.
Example:
s = Askg("Do you like bananas?","Yes/No/Fried only",simple)
if s=="Fried only" print "Impressive"Creating a special chemical dialog for library enumeration.This one is very specialized and is used in combi-chem generator.
Askg( chem_scaffold , enumerate ) → s_makeLib_React_Args
Askg( chem_reaction , enumerate ) → s_makeLib_React_Args
prompts for arguments for
the enumerate library or make reaction commands to create a combinatorial library.
To use this function you need to have the chemical array objects
with Markush-scaffolds or reactions, plus the building blocks
loaded into ICM.
The function returns a string with the agruments for
the enumerate library or make reaction commands.
E.g.
args = Askg( scaff1 enumerate )
enumerate library scaff1 $args
Askg( s_dialogDeclaration ) → "yes"/"no"Generates a dialog from GUI dialog description text. Values from each input field can be accessed either by :
$field_numor
Getarg( i_field_num gui )
buf = "#dialog{\"Select InSilco Models\"}\n"
buf += "#1 l_Passive_GUT_Absorption (yes)\n"
buf += "#2 l_ToxCheck (no)\n"
buf += "#3 l_hERG_QSAR (yes)\n"
buf += "#4 s_Comment_Here ()\n"
Askg(buf)
print $1, $2, Getarg( 3 gui ), $4
Using Askg in shell, html-docs and table tool panels. These variants of the Askg function can also be used as a part of an ICM script in dialogs generated from built-in html documents, or in actions associated with tables.
See also : gui programming
Atan ( real | integer ) - returns the
real
arctangent of its real or integer argument.
Atan ( rarray ) - returns the
rarray of arctangents of rarray elements.
Examples:
print Atan(1.) # equal to 45.
print Atan(1) # the same.
print Atan({-1., 0., 1.}) # returns {-45., 0., 45.} Atan2 ( r_x, r_y ) - returns the
real arctangent of r_y/r_x in the range -180. to 180. degrees
using the signs of both arguments to determine the quadrant of
the returned value.
Atan2 ( R_x R_y ) - returns the
rarray
of arctangents of R_y/R_x elements as described above.
Examples:
print Atan2(1.,-1.) # equal to 135.
print Atan2({-1., 0., 1.},{-0.3, 1., 0.3}) # returns phases {-106.7 0. 73.3} Atanh ( real ) - returns the
real
inverse hyperbolic tangent of its real argument.
Atanh ( rarray ) - returns the
rarray
of inverse hyperbolic tangents of rarray elements.
Examples:
print Atanh(0.) # returns 0.
print Atanh(1.) # returns error
print Atanh({-0.9999, 0., .9999}) # returns { -4.951719, 0., -4.951719 }transforms the input selection to atomic level or returns an atom level selection. Function is necessary since some of the commands/functions require a specific level of selection.
Atom( as|rs|ms|os ) → as_atomLevelSel
Atom( vs ) → as_firstAtomMovedByVar
Atom( as_icmAtom i ) # i-th preceding atom
Atom( as1 [ as_where ] symmetry ) build smiles "C1CCCC1" # a cyclopentane
Atom( a_//c2 symmetry ) # returns 4 other equivalent carbons, c1,c3,c4,c5
#
build string "AFA" # a tripeptide with phenylalanine
Atom( a_/3/ce1 a_/3 symmetry ) # returns ce2 in phe
Atom( as tether )
Atom( vs i ) # i-th preceding atom for variables
Atom( label3d [i_item] ) → as
Atom( pairDist_or_hbondPairDist ) → asmake distance or make bond commands can be used to create distance lines and labels or hbonds, respectively, in the format of a "distance" object;
The Atom function then will return the atoms referenced in the object. E.g. display Atom( hbondpairs ) xstick cpk
Examples:
asel=Acc(a_2/his) # select accessible His residues of
# the second molecule
show Atom(asel) # show atoms of these residues
show Atom( v_//phi ) # carbonyl CsRes, Mol, and Obj functions.
Augment( R_12transformationVector ) - rearranges the transformation vector into an augmented affine 4x4 space transformation
matrix .
The augmented matrix can be presented as
a1 a2 a3 | a4
a5 a6 a7 | a8
a9 a10 a11 | a12
------------+----
0. 0. 0. | 1.M_inv = Power(Augment(R_12direct),-1))
R_12inv = Vector(M_inv)Vector( M_4x4 ) function.
See also:
Vector (the inverse function), symmetry transformations, and transformation vector.
Augment ( R_6Cell ) - returns 4x4
matrix
of oblique transformation from fractional coordinates to absolute coordinates
for given cell parameters {a b c alpha beta gamma}.
This matrix can be used to generate real coordinates. It also contains vectors A, B and C. See also an example.
Example:
read object s_icmhome+"crn.ob"
display a_crn. # load and display crambin: P21 group
obl = Augment(Cell( )) # extract oblique matrix
A = obl[1:3,1] # vectors A, B, C
B = obl[1:3,2]
C = obl[1:3,3]
g1=Grob("cell",Cell( )) # first cell
g2=g1+ (-A) # second cell
display g1 g2 Augment( R_3Vector ) Augment( M_XYZblock ) 1.,1.,..1. column to the Nx3 matrix of with x,y,z coordinates
to allow direct arithmetics with augmented 4x4 space transformation
matrices.
Augment( M_3x3_rotation R_3trans ) 0.,0.,0.,1. row the 3x3 rotation matrix . Then it adds the translation vector as the first three elements of the 4th column.
Axis( { M_33Rot | R_12transformation } ) - returns
rarray with x,y,z components of the normalized rotation/screw axis vector.
Additional information calculated and returned by the function:
r_outrotation angle (in degrees);r_2outhelix rise;R_out3-rarray with a middle point on the axis.
See also:
How to find and display rotation/screw transformation axis
Blob( s_text ['hex'|'base64'] )
Creates blob from string. Hex or Base64 conversion is applied if specified.
Blob( any_variable binary )
Serialize any shell variable into blob
Blob( blob_serialized read )
Un-serialize blob into shell variable.
Example:
read pdb "1crn"
convert auto
make map potential
c = Collection( )
c["ob"] = a_ # store object
c["map"] = m_atoms # store map
s_base64 = String( Blob( c binary ) 'base64') # serialize collection into base64 string.
# now it can be passed between CGI scripts
delete a_*.
delete m_atoms c
c = Blob( Blob( s_base64 'base64' ) read ) # convert s_base64 to blob and un-serialize it
load object c["ob"]
m_atoms = c["map"]
display a_
display m_atoms
Bfactor ( [ as | rs ] [ simple ] ) rarray of b-factors for the specified selection of atoms or residues. If selection of
residue level
is given, the average residue b-factors are returned. B-factors can also be
shown with the command show pdb.
Option
simple returns a normalized b-factor. This option is possible
for X-ray objects containing b-factor information.
The read pdb command calculates the average B-factor for all non-water atoms.
The normalized B-factor is calculated as (b-b_av)/b_av .
This is preferable for coloring ribbons by B-factor since these numbers
only depend on the ratios to the average.
We recommend to use the following commands to color by b-factor:
color ribbon a_/ Trim(Bfactor( a_/ simple ),-0.5,3.)//-0.5//3. # or
color a_// Trim(Bfactor( a_// simple ),-0.5,3.)//-0.5//3. # for atomsSee also: set bfactor.
Examples:
read pdb "1crn"
avB=Min(Bfactor(a_//ca)) # minimal B-factor of Ca-atoms
show Bfactor(a_//!h*) # array of B-factors of heavy atoms
color a_//* Bfactor(a_//*) # color previously displayed atoms
# according to their B-factor
color ribbon a_/A Bfactor(a_/A) # color the whole residue by mean B-fac.real Boltzmann constant = 0.001987 kcal/deg.
Example:
deltaE = Boltzmann*temperature # energygrob,
and interactively moved and resized with the mouse.
One can select atoms inside a box by this operation:
as_ & Box( )
Box ( [ display ] ) rarray with {Xmin ,Ymin ,Zmin ,Xmax ,Ymax ,Zmax }
parameters of the graphics box as defined on the screen. With the display keyword,
the function returns {0. 0. 0. 0. 0. 0.} if the box is not displayed (by default
it returns the last 6 values).
Box ( center ) rarray with Xcenter,Ycenter,Zcenter,Xsize,Ysize,Zsize
parameters of the graphics box as defined on the screen.
Box ( as [ r_margin ] ) rarray
with Xmin,Ymin,Zmin,Xmax,Ymax,Zmax parameters of the box surrounding
the selected atoms. The boundaries are expanded by r_margin
(default: 0.0 ).
Examples:
build string "se ala his" # a peptide
display box Box(a_/2 1.2) # surround the a_/2 by a box with 1.2A margin
color a_//* & Box( ) Box ( { g | m | R_6box } [ r_margin ] ) - returns the 6-
rarray with Xmin,Ymin,Zmin,Xmax,Ymax,Zmax parameters of the box surrounding
the selected grob or map. The boundaries are expanded by r_margin
(default: 0.0 ).
Bracket ( m_grid [ r_vmin r_vmax ] ) - returns the truncated
map . The map will be truncated by value. The values beyond
r_vmin and r_vmax will be set to r_vmin and r_vmax
respectively.
Bracket ( m_grid [ R_6box ] ) - returns the modified
map . All the values beyond the specified box will be
set to zero. Example:
make map potential "gh,gc,gb,ge,gs" a_1 Box()
m_ge = Bracket(m_ge, Box( a_1/15:18,33:47 )) # redefine m_geSee also:
Rmsd( map ) and Mean( map ), Min( map ), Max( map ) functions.
Rmsd, is contact based and can measure the difference
in a wide range of model accuracies. Roughly speaking it measures the
surface weighted fraction of native contacts. Can be used to evaluate
the differences between several NMR models, the accuracy of models by
homology and the accuracy of docking solutions.
Cad can measure the geometrical difference between two conformations in several different ways:
- between two conformations of the same protein based on full atom
residue-residue contact area calculation,
Cad(..) - between two conformations of the same protein based on Cbeta-Cbeta
distance evaluation (`Cad1{Cad}(..
distance) .ICM uses an empirically derived ContactStrength( Cb-distance ) function. - between two homologous structures based preservation of the residue
contacts through the alignment ( Cad (..
alignment)) . The contact strength in this case is also derived from the inter-residue distances.
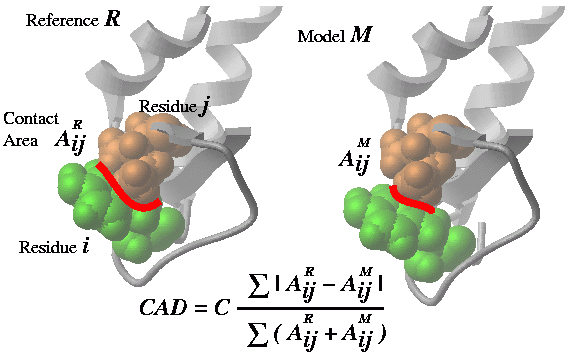
Cad ( rs_A1 [ rs_A2] rs_B1 [ rs_B2] [ distance | alignment ] ) - returns the
real contact area difference measure (described in
Abagyan and Totrov, 1997)
between two conformations A and B of the same set of residue pairs
from two different objects.
The set of residue pairs in each object (A or B) can be defined in two ways:
- by a single selection rs_A1 : all pairs between selected residues (is equivalent to rs_A1 rs_A1 )
- by two residue selections rs_A1 rs_A2: cross pairs between two sets of selected residues (e.g. the contacts between two subunits)
The whole matrix of contact area differences is returned in
M_out . This matrix can be nicely plotted with the
plot area M_out number ..
command (see example). The full matrix can also be
used to calculate the residue profile of the differences.
See also:
Area() function which calculates absolute residue-residue contact areas.
Options:
distanceoption allows one to compare approximations of the inter-residue contact areas by the Ca and Cbeta positions. This allows one to calculated deformations between two homologous proteins which is not possible in the default mode in which two chemically identical molecules are compared. The residue pairs in two homologs are equivalenced according to the alignments linked to the molecules. Residues deleted in a homologue are considered to have zero contact.alignmentoption is described in Marsden, Abagyan, 2004, Bioinformatics, v20, 2333-2344.
Examples:
# Ab initio structure prediction, Overall models by homology
read pdb "cnf1" # one conformation of a protein
read pdb "cnf2" # another conformation of the same protein
show 1.8*Cad(a_1. a_2.) # CAD=0. - identical; =100. different
show 1.8*Cad(a_1.1 a_2.1) # CAD between the 1st molecules (domains)
show 1.8*Cad(a_1.1/2:10 a_2.1/2:10) # CAD in a window
PLOT.rainbowStyle = 2
plot area grid M_out comment=String(Sequence(a_1,2.1)) link display
# Loop prediction: 0% - identical; ~100% totally different
# CAD for loop 10:20 and its interactions with the environment
show 1.8*Cad(a_1.1/10:20 a_1.1/* a_2.1/10:20 a_2.1/*)
# CAD for loop 10:20 itself
show 1.8*Cad(a_1.1/10:20 a_1.1/10:20 a_2.1/10:20 a_2.1/10:20)
# Evaluation of docking solutions: 0% - identical; 100% totally different
read pdb "expr" # one conformation of a complex
read pdb "pred" # another conformation of the same complex
show Cad(a_1.1 a_1.2 a_2.1 a_2.2) # CAD between two docking solutions
#
# ANOTHER EXAMPLE: the most changed contacts
read object "crn"
copy a_ "crn2"
randomize v_ 5.
Cad(a_1. a_2.)
show s_out
read column group input= s_out name="cont"
sort cont.1
show cont
# the table looks like this (the diffs can be both + and -):
#>T cont
#>-1-----------2-----------3----------
-39. a_crn.m/38 a_crn.m/1
-36.4 a_crn.m/46 a_crn.m/4
-32.1 a_crn.m/46 a_crn.m/5
-29.8 a_crn.m/30 a_crn.m/9
-25.2 a_crn.m/37 a_crn.m/1
...
42.5 a_crn.m/43 a_crn.m/5
45.1 a_crn.m/44 a_crn.m/6
45.2 a_crn.m/43 a_crn.m/6
55.3 a_crn.m/46 a_crn.m/7
56. a_crn.m/45 a_crn.m/7 Cad ( rs_A1 [ rs_A2] rs_B1 [ rs_B2] alignment ) Ceil ( r_real [ r_base] ) - returns the smallest
real
multiple of r_base exceeding r_real.
Ceil ( R_real [ r_base] ) - returns the
rarray
of the smallest multiples of r_base exceeding components of the
input array R_real. Default r_base= 1.0 .
See also:
Floor( ).
Cell ( { os | m_map } ) - returns the
rarray
with 6 cell parameters {a,b,c,alpha,beta,gamma} which were assigned to the
object or the map.
Charges can also be shown with a regular show as_select command.
Charge ( { os | ms | rs | as } [ formal | mmff ] ) - returns
rarray of elementary or total charges
depending on the selection level.
formal: return formal chargesmmff: return formal charges calculated according to mmff atom types and rules. Note: do not confuse this option with a function to return the mmff charges.
Examples:
build string "ala his glu lys arg asp"
show Charge(a_1) # charge per molecule
show Charge( a_1/* ) # charge per residue
show Charge( a_1//* ) # charge per atom
avC=Charge(a_/5) # total electric charge of 15th residue
avC=Sum(Charge(a_/5/*)) # another way to calculate it
show Charge(a_//o*) # array of oxygen charges
# to return mmff charges:
set type mmff
set charge mmff
Charge( a_//* )
# to return total charges per molecular object:
read mol s_icmhome+"ex_mol.mol"
set type mmff
set charge mmff
Charge( a_*. )Converting 3D objects to chemical arrays.
Chemical( ms|os [exact] [hydrogen] [unique] [pharmacophore] [stack] )
returns an array of chemicals from a molecular selection of 3D molecular objects, e.g. a_H for hetero-molecules
By default the selected molecules will be converted to 2D graphs. However with
the exact option the original 3D coordinates will be retained in the elements of the chemical array.
If you want to preserve explicitly drawn hydrogens hydrogen option should be used.
Note that the number of chemicals in the array will be determined by the selection level. At the object
level multiple molecules of the same object will be merged into one array element.
With unique option duplicates will be excluded from the result.
If 3D object has an embedded conformation stack you can use stack option to preserve it in the result chemical object
Example:
read pdb "1ch8"
group table t_2D Chemical(a_H) # convert to 2D chemical table
group table t_3D Chemical(a_H exact) # make 3D chemical table without hydrogens
group table t_3D_hyd Chemical(a_H exact hydrogen) # make 3D chemical table, keep hydrogens
With pharmacophore option the function generates pharmacophore points for the input selection.
Example:
read object s_icmhome + "biotin.ob" name="biotin"
read mol input = String( Chemical(a_ pharmacophore )) name="biotin_ph4"
display xstick
display wire a_biotin.
To display supported pharmacophore types and use show pharmacophore type command
Converting smiles to chemical arrays:
Chemical( S_smiles|s_smiles )returns an array of chemicals from a string arrays of smiles.
Example:
add column t Chemical({"N[C@@](F)(C)C(=O)O", "C[C@H]1CCCCO1"})Converting InChI to chemical arrays:
Chemical( S_InChI|s_InChI )
See also: chemical functions
Generating combinatorial compounds from a Markush structure and R-group arrays.
Chemical( scaffold I_RgroupNumArray enumerate ) → returns one chemicalThe I_RgroupNumArray is an array of as many elements as there are different R groups in the scaffold.E.g. if there is R1 R2 R3 than this parameter can be {10,21,8}. The numbers refer to the R-group arrays linked to the scaffold.E.g.
group table scfld Chemical("C(=CC(=C(C1)[R2])[R1])C=1") "mol"
link group scfld.mol 1 Chemical({"N","O","S"})
link group scfld.mol 2 Chemical({"[Cl]", "[C*](=O)O"})
Nof( scfld.mol library ) # returns the total number of molecules in that combinatorial library
Nof( scfld.mol group ) # returns an array of sizes of each linked array in R1 R2.. order.
Chemical( scfld.mol {1 1} enumerate )
Chemical( scfld.mol {1 2} enumerate )
Chemical( scfld.mol {2 2} enumerate )
Chemical( enumerate scaffold [simple] R1 R2 ... ) → returns enumeration result
The same as above but does not require explicit linkage with link group command.
Example:
Chemical( enumerate Chemical("C(=CC(=C(C1)[R2])[R1])C=1") Chemical({"N","O","S"}) Chemical({"[Cl]", "[C*](=O)O"}) )
simple mode is similar to enumerate library and requires that size of R-group arrays be the same.
Example:
Chemical( enumerate Chemical("C(=CC(=C(C1)[R2])[R1])C=1") simple Chemical({"N","O"}) Chemical({"[Cl]", "[C*](=O)O"}) )See also: linking scaffold to R-group arrays and the Nof
[ Collection ]
Cluster( I_NxM_NearestNeighb i_M_totalNofNearNeighbors i_minNofCommonNeighbors ) → I_N_clusterNumbersfunction returns
iarray of cluster numbers for each or N points.
The input to the first function is an array of M nearest neighbors (defined by the second argument i_M_totalNofNearNeighbors) for each of N points. For example for an array for 5 points, and i_M_totalNofNearNeighbors = 3 it can be an array like this:
{3,4,5, 1,3,4 1,2,5 2,3,5 1,2,3} . The points will be grouped into
the same cluster if the number of neighbors they share is larger or equal than i_minNofCommonNeighbors .
This clustering algorithm is adaptive to the cluster density
and does not depend on absolute distance threshold. In other words it will identify both very sparse clusters
and very dense ones. The nearest neighbor array can be calculated by the
with the Link( I_bitkeys , nBits, nNearestNeighbors ) function.
Cluster( M_NxNdist r_maxDist ) → I_N_clusterNumbers This function identifies the i_totalNofNeighbors nearest neighbors from the full distance matrix M_NxNdist for each point and assembles points sharing the specified number of common neighbors in clusters.
All singlets (a single item not in any cluster) are placed in a special cluster number 0 . Other items are assigned to a cluster starting from 1.
Example with a distance matrix:
# let us make a distance matrix D
# we will cook it from 5 vectors {0. 0. 0.}
m=Matrix(5,3) # initialize 5 vectors
m[2,1:3]={1. 0. 0.} # v2
m[3,1:3]={1. 1. 0.} # v3
m[4,1:3]={1. 1. 1.} # v4
m[5,1:3]={1. 0.1 0.1} # v5 close to v2
D = Distance( m ) # 5x5 distance matrix created
Cluster( D , 0.2 ) # v2 and v5 are assigned to cluster 1
Cluster( D , 0.1 ) # radius too small. All items are singlets
Cluster( D , 2. ) # radius too large. All items are in cluster 1
The function to create a collection object
Collection()collection object
Collection( s_json_string )collection object from a text in JSON format
Collection( s_url_encoded_string )collection object from a URL encoded string ("a=1&b=abc")
Collection( web )collection object from the POST or GET arguments. Can be used in CGI scripts. Multi-part content is also supported.
Collection( S_uniq_names_n S_values_n ) Replace( S_name_array k_translation_collection ).
Collection( table_row )collection object for the table row. Collection( t[1] )
Collection( table {column|header|all} )collection
Collection( table|tab_column format )collection object with the members controlling format, color and function for calculated columns. This collection can be modified and set back to the table or table column with the set format collection command . Example:
add column t {1 2 3} {1 2 3}
add column t function= "A+B"
set format t.A "<i>%1</i>"
show format t
c = Collection(t.A format ) # modify c
set format t.B c
[ Color from gradient | Color image | Color protein ]
returns RGB values or color names. Summary:- : Color(
[ball|cpk|label|skin|surface|wire|ribbon|label] ) => M_Nx3_rgb - : Color( background ) =>
- : Color(
) => M_Nx3_rgb - : Color(
[ ] ) => S_system_color_names_or_hex - : Color(
) => S_n_colorNames from _red to _black - : Color(
[ ] ) => R_3_rgb|M_Nx3_rgb # interpolation - : Color(
- : Color(
[name] ) => - : Color(
background ) => - : Color(
[ [ ]] ) => - : Color( background ) =>
Color ( as_n ball|cpk|label|skin|surface|wire ) → M_nx3_rgbmatrix of colors for a particular representation (0. 0. 0. 0. means black or undisplayed )
Color ( g_grob ) → M_nx3_rgbmatrix of RGB numbers for each vertex of the g_grob (dimensions: Nof ( g_grob),3).
See also:
color grob matrix .
Example:
build string "se his"
display xstick
make grob image name="g_"
display g_ only smooth
M_clr = Color( g_ )
for i=1,20 # shineStyle = "color" makes it disappear completely
color g_ (1.-i/20.)*M_clr
endfor
color g_ M_clr Color( M_rgb [name] ) → S_colorHex_or_Names
- returns sarray of color names in hex code, or, with option name , ICM colors approximating the rgb values in the matrix.
The ICM color names and definitions are taken from the icm.clr file.
Example:
m = Matrix(3)
Color(m ) # returns {"#ff0000","#00ff00","#0000ff"}
Color(m name) # returns icm approximations {"red","lime","midnightblue"}
Color( system )
- returns sarray of system color names.
Color( system i_numColor ) - returns a name of a system color by number.
Example:
N = Nof( Color( system ))
for i=1,10
print Color( system Random(N) ) # randomly pick one color
endfor
Color( background ) - returns
rarray of three RGB components of the background color.
Color( r_value s_gradient [ r_from r_to ] ) → R_3rgbcolor .. rgb= command.
Color( R_N_values s_gradient [ r_from r_to ] )
Note that these colors are from the permanent part of the spectrum and are only approximately equal to transient colors resulting from the color-by-rainbow-and-value command like
GRAPHICS.NtoCRainbow = "white/lightpink/red/darkred"
R = Bfactor(a_/* )
color a_/* R//0.//100. # uses a perfect rainbow at a transient part of the spectrum
Color( R "white/lightpink/red/darkred" 0. 100. ) # projects those colors to the 'named' part of the spectrumExamples:
s = "red/lime/blue"
Color( 0. s 0. 1. )
Color( 0.5 s 0. 1. )
Color( 1.0 s 0. 1. )
Color( 0.1 s 0. 1. )
Color( 0.8 s 0. 1. )
Color( {0.1 0.8} s 0. 1. )
Color( {1. 8.} s 0. 10. )
Color( 0.1 "red/lime/blue,0:1" )
Color( {0.1 0.8} "red/lime/blue,0:1" )
Color( {1. 8.} "red/lime/blue,0:10" )
Color( imageArray background )
returns sarray with background colors of the images in imageArray_.The color of the top left pixel of the image is returned as the background color currently.
See also: Image, image parray
Color( S|s_aa protein ) → S|s_aaHexColorsSome tables may contain an amino acid (along with its position) in its cell, e.g. one may record amino acids around the binding site:
add column t "D12"//"E13"//"K14" "D"//"A"//"L"
show Color(t.A protein ) # returns sarray of colors for each value
show Color(t.A[1] protein ) # returns string color, eg #AAFFFF for "D12"
set format t.A color='Icm::Color(A protein)' # will color cells in the table.
See also: set format
Consensus ( ali ) → s_consensus- returns the
string consensus of alignment ali_.
The
consensus characters are these:
# hydrophobic; + RK; - DE; ^ ASGS; % FYW; ~ polar.
In the
selections by consensus
a letter code (h,o,n,s,p,a) is used.
Consensus ( ali { i_seq | seq } ) - returns the
string
consensus of alignment ali_ as projected to the sequence.
Sequence can be specified by its order number in the alignment or by name.
Example displaying conserved residues:
read alignment "sx" # load alignment
read pdb "x" # structure
display ribbon
# multiply rs_ by a mask like " A C N .."
cnrv = a_/A & Replace(Consensus(sx cd59),"[.^~#]"," ")
display cnrv red
display residue label cnrv
Consensus ( ms|rs ) surface accessible areas projected on the selected residues via linked sequence and alignment.
making a table with the contour lines of a 2D function represented by a matrix for display in the plot command.
Contour( M [r_step|i_numContours [fmin,fmax]] [R_Xs|R_Ys] ) → T_contourData (X,Y,conn,Z)Example (UNFINISHED):
M = 10.*Smooth(Smooth(Smooth(Matrix(100))))
tt = Contour(M,10,0.,5.)
delete tt.Z == 0.
sort tt.Z
add column tt "_black line 0.5" name="mark"
plot tt.X tt.Y tt.mark "/tmp/tmp.eps" append
Corr ( R_X, R_Y ) → r_correlation- returns the
real value of the linear correlation coefficient.
Probability of the null hypothesis of zero correlation is stored in r_out .
Note: this function returns R , not R2 .
Taking it to the 2nd power can be a humbling experience.
Examples:
r=Corr(a,b) # two vectors a and b
if (Abs(r_out) < 0.3) print "it is actually as good as no correlation"LinearFit( ) function.
Cos ( { r_Angle | i_Angle } ) - returns the
real
value of cosine of its real or integer argument.
Cos ( rarray ) - returns
rarray
of cosines of each component of the array.
Examples:
show Cos(60.) # returns 0.5
show Cos(60) # the same
rho={3.2 1.4 2.3} # structure factors
phi={60. 30. 180.} # phases
show rho phi rho*Cos(phi) rho*Sin(phi) # show in columns rho, phi,
# Re, Im Cosh ( { r_Angle | i_Angle } ) real value of hyperbolic cosine of its real or integer argument.
Cos(x)=0.5( eiz + e-iz )
Cosh ( rarray ) rarray of hyperbolic cosines of each component of the array.
Examples:
show Cosh(1.) # 1.543081
show Cosh(1) # the same
show Cosh({-1., 0., 1.}) # returns {1.543081, 1., 1.543081}-
Count( i_n ) → I_1,2,3,..n -
Count( i_from i_to [i_step=1] ) → I_from,...,to -
Count( I|R|S_array ) → I_1,..,n -
Count( I|R|S unique|identity|number) → I # 111222233 or 123123412 or 333444422
Detailed descriptions:
Count ( [ i_Min, ] i_Max ) iarray of numbers growing from i_Min to i_Max.
The default value of i_Min is 1.
Examples:
show Count(-2,1) # returns {-2,-1,0,1}
show Count(4) # returns {1,2,3,4}Iarray( ).
Count ( array ) - returns
iarray
of numbers growing from 1 to the number of elements in the array.
Count ( I|R|S_array unique | identity ) → Ireturns an integer array with integer id for sequentially identical values. Example:
group table t {"d","d","d","bb","bb","a","a","a"}
add column t Count(t.A unique ) Count(t.A identity ) name={ "unique","identity" }
show t
#>T t
#>-A-----------unique------identity---
d 1 1
d 1 2
d 1 3
bb 2 1
bb 2 2
a 3 1
a 3 2
a 3 3
CubicRoot( r ) → r_cubic_root
CubicRoot( r [ r_im ] ) → R6_3re+3imExample:
CubicRoot(27. )
3.
CubicRoot(27. 0.)
#>R
3.
-1.5
-1.5
0.
-2.598076
2.598076
See also: SolveCubic, Sqrt
Summary:
Date( ) → e_1currentDate
Date ( filename modify ) → e_dateOfLastFileModification
Date( n ) → e_arrayOf_n_currentDates
returns an date array of current system date and time. It may be used as a column in a table.
Example:
print "Today is :" Date()
add column t Count(10) Date(10)
t.B[1] = Date() # today
t.B[2] = Date()-1 # yesterday
t.B[3] = Date()+1 # tomorrow
Date ( version ) → e_dateOfCompilation
Date ( os ) → e_pdbDates
returns the date of the pdb file creation in an date array format.
The date read from the HEADER record of a pdb file and is stored with the object.
Example:
read pdb "1crn"
if Date(a_) > Date("1980","%Y") print "released after 1980"
Date ( {s_date_string|S_dates} [ s_format_to_parse_date_string ] )
converts string or sarray to dates using s_format or default TOOLS.dateFormat and always returns a parray of dates. If the first argument is a string, it will have one element in the array.
Example:
String( Date( "12 Oct 2002", "%d %b %Y" ) "%Y-%m-%d" )The allowed format specifications are the following:
| format | description |
|---|---|
| %a or %A | The weekday name according to the current locale, in abbreviated form or the full name. |
| %b or %B | The month name according to the current locale, in abbreviated form or the full name. |
| %c | The date and time representation for the current locale. |
| %C | The century number (0-99). |
| %d or %e | The day of month (1-31). |
| %D | Equivalent to %m/%d/%y. (This is the American style date, very confusing to non-Americans, especially since %d/%m/%y is widely used in Europe.) |
| %H | The hour (0-23). |
| %I | The hour on a 12-hour clock (1-12). |
| %j | The day number in the year (1-366). |
| %m | The month number (1-12). |
| %M | The minute (0-59). |
| %n | Arbitrary whitespace. |
| %p | The locale’s equivalent of AM or PM. (Note: there may be none.) |
| %r | The 12-hour clock time (using the locale’s AM or PM). (%I:%M:%S %p) |
| %R | Equivalent to %H:%M. |
| %S | The second (0-60; 60 may occur for leap seconds; earlier also 61 was allowed). |
| %t | Arbitrary whitespace. |
| %T | Equivalent to %H:%M:%S. |
| %U | The week number with Sunday the first day of the week (0-53). The first Sunday of January is the first day of week 1. |
| %w | The weekday number (0-6) with Sunday = 0. |
| %W | The week number with Monday the first day of the week (0-53). The first Monday of January is the first day of week 1. |
| %x | The date, using the locale’s date format. |
| %X | The time, using the locale’s time format. |
| %y | The year within century (0-99). When a century is not otherwise specified, values in the range 69-99 refer to years in the twentieth century (1969-1999); values in the range 00-68 refer to years in the twenty-first century (2000-2068). |
| %Y | The year, including century (for example, 1991). |
Example:
String( Date() "%b %d %Y %I:%M%p" ) # Current date and time in American style
String( Date() "%d/%b/%Y %H:%M" ) # European style Deletion ( rs_Fragment, ali_Alignment [, seq_fromAli ] [, i_addFlanks ] [{"all"|"nter"|"cter"|"loop"}] ) - returns the
residue selection
which flanks deletion points from the viewpoint of other sequences in the
ali_Alignment. If argument seq_fromAli is given (it must be the
name of a sequence from the alignment), all the other sequences in the alignment
will be ignored and only the pairwise sub-alignment of rs_Fragment and
seq_fromAli will be considered. The alignment must be
linked
to the object. With this function (see also Insertion function)
one can easily and quickly visualize and/or extract all indels
in the three-dimensional structure. The default i_addFlanks
parameter is 1. String options:
- "all" (default: no string option) select deletions of all types
- "nter" select only N-terminal fragments
- "cter" select only C-terminal fragments
- "loop" select only the internal zones of deleted loops
See example coming with the
Insertion( )
function description.
Descriptor ( chemArray number )
- returns table of various numerical descriptors of the following categories:
# https://www.epa.gov/sites/production/files/2015-05/documents/moleculardescriptorsguide-v102.pdf
- Atom counts
- a_count - total atom count
- a_heavy - heavy atom count
- a_aro - aromatic atom count
- HB_a - hydrogen bond acceptor count
- HB_don - hydrogen bond donor count
- Individual atom counts
- a_nH a_nC a_nN a_nO a_nF a_nP a_nS a_nCl a_nBr
- Bond counts
- b_count - total number of bonds
- b_heavy - number of bonds between heavy atoms
- b_rotN - number of non-ring bonds
- b_rotR - fraction of non-ring bonds
- b_1rotN - number of single non-ring bonds
- b_1rotR - fraction of single non-ring bonds
- b_ar - number of aromatic bonds
- b_single - number of single bonds
- b_double - number of double bonds
- b_triple - number of triple bonds
- b_rot - number of aromatic bonds
- Topological Descriptors ([Hall 1991] and [Hall 1997])
- Definitions
- n[i] : atomic number of atom i
- d[i] : number of heavy connections for atom i
- v[i] : nV[i] - nH[i] for atoms with atomic number <= 10, (nV[i] - nH[i]) / (n[i] - nV[i] - 1) for atoms with atomic number > 10
- P[i] : number of paths of bond length i in hydrogen suppressed molecule
- N : number of non-hydrogen atoms
- R[i] : van der Waals radius of atom i
- Rc : van der Waals radius of carbon atom
- α : 1 - Sum( R[i] / Rc )
- Na : N + α
- Pa[i] : P[i] + α
- Chi Connectivity Indices
- t_chi0 : Atomic connectivity index order 0. Sum(1/sqrt(d[i]))
- t_chi0_C : Carbon connectivity index order 0. (Same as above calculated for carbon atoms)
- t_chi1 : Atomic connectivity index order 1. Sum(1/sqrt(d[i]*d[j])) for all bonds between heavy atoms i and j.
- t_chi1_C : Carbon connectivity index order 1. (Same as above calculated for carbon atoms)
- t_chi0v : Atomic valence connectivity index order 0. Sum(1/sqrt(v[i]))
- t_chi0v_C : Carbon valence connectivity index order 0. (Same as above calculated for carbon atoms)
- t_chi1v : Atomic valence connectivity index order 1. Sum(1/sqrt(v[i]*v[j])) for all bonds between heavy atoms i and j.
- t_chi1v_C : Atomic valence connectivity index order 1. (Same as above calculated for carbon atoms)
- Kappa Shape Indices
- k1 : First kappa shape index: N(N-1)^2 / P[1]^2
- k2 : Second kappa shape index: (N-1)(N-2)^2 / P[2]^2
- k3 : Third kappa shape index: (N-1)(N-3)^2 / P[3]^3 for odd N, (N-3)(N-2)^2 / P[3]^2 for even N
- k1a : First kappa shape index α: Na(Na-1)^2 / Pa[1]^2
- k2a : Second kappa shape index: (Na-1)(Na-2)^2 / Pa[2]^2
- k3a : Third kappa shape index: (Na-1)(Na-3)^2 / Pa[3]^3 for odd N, (Na-3)(Na-2)^2 / Pa[3]^2 for even N
- zagreb : Sum(d[i]*d[i]) for all non-hydrogen atoms
- weiner : Sum(D(i,j)) for all non-hydrogen atom pairs i,~~j. D(i,j) - topological distance between atoms i and j.
- maxdist : Max(D(i,j)) for all atom pairs i,~~j. D(i,j) - topological distance between atoms i and j.
- Definitions
Example:
read table s_icmhome + "celebrex50.sdf" name="t"
add column t Descriptor( t.mol number )
Descriptor ( chemArray )
Descriptor ( chemArray collection_of_Fingerprint_Parameters [info] ) - returns vector of binary fingerprints, default or custom, calculated for each chemical.
The collection_of_Fingerprint_Parameters argument is a collection which defines parameters for fingerprint generation and consists of the following members:
- ATMAP: string with comma separate atom
propertiesdescriptions. Example: cd,h - BOMAP: string with comma separate bond
propertiesdescription. Example: bt,r - SIZE : result vector size
- LEN : maximum fragment/chain length
- TYPE : fingerprint type: "linear","triplets","ecfp"
- BINARY: yes/no (no - counted fingerprint mode)
- ECFPITER: (ecfp only) number of iterations. 1 - ECFP2, 2 - ECFP3, 3 - ECFP4, etc...
Examples:
# export default binary fingerprints
write sarray Sarray(Descriptor(t.mol)) name="fp.txt"# linear counted fingerprint, SIZE=1024, max chain length=5
Descriptor( t.mol, Collection("ATMAP" "cd,h" "SIZE" 1024 "BOMAP" "bt,r" "LEN" 5 "TYPE" "linear", "BINARY", no) )
# ecfp4 fingerprint
Descriptor( t.mol, Collection("ATMAP" "cd,h" "SIZE" 2048 "BOMAP" "bt" "LEN" 999 "TYPE" "ecfp", "ECFPITER" 3, "BINARY", no) )Example in which fingerprints returned by the function are directly used in distance calculation:
add column t Chemical({"CCN","CCCN"})
mod = Collection()
mod['FP'] = Descriptor( t.mol )
Distance(mod['FP'], Descriptor(Chemical("C(=O)CCN")))
Descriptor ( chemArray predModel )
- returns vector of rarrays with chemical descriptors calculated for each chemical.
each rarray consists of chemical fingerprint part and values for columns with formula used in the predModel.
This information can be used for further analysis or exported outside ICM.
Example:
# assumes that 'clogPpred' is a prediction model
tt = Table( Transpose( Matrix( Descriptor( Chemical("CCC"), clogPpred ) ) ))
add column tt Name( clogPpred column )
sort reverse tt.ATo find the description of the each particular position in the rarray Name function can be used.
Example:
rr = Descriptor( Chemical("CCC") myModel )[1]
na = Name( myModel column )
for i=1,Nof(rr)
if (rr[i] != 0) print rr[i], na[i]
endfor
See also: Name( predModel, column .
Det ( matrix ) - returns a
real determinant of specified square matrix.
Examples:
a=Rot({0. 0. 1.}, 30.) # Z-rotation matrix by 30 degrees
print Det(a) # naturally, it is equal to 1.group sequence se1 se2 se2 se4 mySeqs
align mySeqs
distMatr=Distances(mySeqs)or any objects between which one can somehow define pairwise distances. Since principal coordinates are sorted according to their contribution to the distances and we can hardly visualize distributions in more than three dimensions, the first two or three coordinates give the best representation of how the points are spread in n-1 dimensions. Another application is restoring atomic coordinates from pairwise distances taken from NMR experiments.
Disgeo ( matrix ) - returns
matrix
[1:n,1:n] where the each row consists of n-1 coordinates of point
[i] sorted according to the eigenvalue (hence, their importance).
The first two columns, therefore, contain the two most significant
coordinates (say X and Y) for each of n points.
The last number in each row is the eigenvalue [i].
If distances are Euclidean, all the eigenvalues are positive or
equal to zero. The eigenvalue represents the "principal coordinate"
or "dimension" and the actual value is a fraction of data
variation due to the this particular dimension. Negative
eigenvalues represent "non-Euclidean error" in the initial distances.
R_outreturns four numbers: total negative eigen values, and the first 3 largest positive eigenvalues. All scaled to 100%.
Example:
read sequences s_icmhome+"zincFing" # read sequences from the file,
list sequences # see them, then ...
group sequence alZnFing # group them, then ...
align alZnFing # align them, then ...
a=Distance(alZnFing) # a matrix of pairwise distances
n=Nof(a) # number of points
b=Disgeo(a) # calculate principal components
corMat=b[1:n,1:n-1] # coordinate matrix [n,n-1] of n points
eigenV=b[1:n,n] # vector with n sorted eigenvalues
xplot= corMat[1:n,1]
yplot= corMat[1:n,2]
plot xplot yplot CIRCLE display # call plot a 2D distribution[ Distance iarray | Distance rarray | Distance ~~as_ | Distance ~~as_ rarray | Distance matrix | Distance hierarchical | Distance Tanimoto | Distance chemset | Distance 2 matrices | Distance tether | Distance Dayhoff | Distance in alignment | Distance 2 alignments | Distance tree | Distance chemical ]
generic distance function. Calculates distances between two ICM-shell objects, bit-strings or molecular objects, or extracts distances from complex ICM-shell objects.
Distance( II | RR | as as | seq seq ) → r_dist
Distance( S|s, s) → R|r
Distance( ali ali [exact] ) → r
Distance( S S [simple]) → M
Distance( Sn_hier_codes Sm_hier_codes tree [ s_delimeter ]) → M_nm_0to1
Distance( Mnk ) → Mnn_cart_dist_between_row-vectors
Distance( Mnk Mmk ) → Mnm
Distance( M_xyz|as M_xyz|as r_dist ) → l_yes_if_closer_than_dist
Distance( seq seq [identity|evolution|new|fast|number|reverse] )
Distance( seq seq nucleotide [len] )
Distance( seqArr[n]> ) → <M_nn
Distance( ali seq [string] ) → R_n_seq_in_ali
Distance( seqArr[n]> <seq ) → R_n
Distance( seqArr[n]> <seqArr[m]> ) → <M_nm
Distance( as [r_default=-1.] ) → R_tether_lengths_or_def
Distance( as_n as_m ) → d_between_centers_of_mass
Distance( as_n as_m all ) → R_nm
Distance( as_n as_n rarray ) → R_n # aligned arrays, same n
Distance( ali [0] ) → M_interSeqDist
Distance( X_n [X_m] [pharmacophore|sstructure] ) → M_nxm_chemical_Tanimoto_distances
Distance( X_n X_m [[R_Wn R_Wm] r_maxdist (0.4) [r_steepness(6.)]] set ) → r_set1_2_distance
Distance( bitvecArr[n]> <bitvecArr[m]> ) → <M_nm #tanimoto, see Descriptor function
Distance( I_keys1 I_keys2 i_nBits|R_nbitWeights [simple] ) → M : Tanimoto|weighted
Distance( tree [i_at=1] split ) → r_splitLevel
Distance( tree all|modify ) → R_splitLevels|splitLevelTStats
Distance( g wire|grid [i_maxDist(1000000)>] ) → <M_shortestPaths
Distance( d_0to1|M evolution ) → d|M_Dayhoff_correction_applied
See detailed descriptions below.
Distance ( iarray1, iarray2 ) - returns the
real sqrt of sum of (I1i -I2i )2 .
Distance ( R_X, R_Y ) real Cartesian distance between two vectors of the same length.
D = Sum( ( Xi - Yi )2 )
Distance ( as_1, as_2 [ all ] )
- returns the real distance in Angstroms between centers of mass of
the two specified selections. The interactive usage of this function:
Option all will return an array of all cross distances between the selections.
The selected virtual atoms will be skipped if the selection level residue, molecule or object.
Othewise, if you explicitly select virtual atoms, they will be included, e.g.
build string "ala" # contains 2 virtual atoms at N terminus
build string "his" # also contains 2 virtual atoms at N terminus
Distance( a_1. a_2. all ) # no virtual atom distances
Distance( a_1.// a_2.// all ) # selected virtual atoms are included
Distance( a_1. a_2. ) # a single distance between centers of mass Distance ( as_1 , as_2, rarray )
- returns the rarray of distances in Angstroms between the two specified selections containing
the same number of atoms (1-1, 2-2, 3-3, ...).
See also: Distance ( as1 as2 all )
Distance ( M_coor ) matrix
of distances between the rows of the input matrix M_coor.
Each row contains m coordinates (3 in 3D space). For example:
Distance(Xyz(a_//ca)) returns a square matrix of Ca-Ca distances.
Distance( Sn_hier_codes Sm_hier_codes tree [s_delimeter]) → M_nm compares two arrays of hierarchical labels like this: "clan.family.subfamily.." The labels can be delimiter separated, or just strings of the same length where one positions is just one character (the default). Returns a distance matrix normalized to [0:1] range.
Here are examples of classification codes that can be used:
- EC numbers, enzymes: eg Distance({"6.2.1.11"},{"6.2.1.25"}, tree, ".")
- ATC drug codes, eg Distance({"C-03-C-C-01"},{"L-01-A-B-03"}, tree, "-") , use
Replaceto insert dashes between sections - AHFS drug codes, eg Distance({"08:18.32"},{"08:16.00","92:00.00"}, tree, ":.")
- ...
Examples:
add column t {"Cocaine","Betaxolol"} {"52:16.00","52:92.00"}
show Distance(t.B t.B tree ":.")
add column tt {"Cocaine","Betaxolol"} {"AB2","ABc"}
show Distance(tt.B tt.B tree ) # use each character as level
These distance matrices can be used for making 2D and 3D graphs, or to plot clustering trees of tables containint suitable label columns, eg see ds3D make tree object .
Distance( X_chem_n X_chem_m ) → M_nxm_distances
Distance( I_keys1 I_keys2 nBits | R_nBitWeights [simple] ) → M_distances - returns the
matrix of Tanimoto distances between two arrays of bit-strings.
Each array of N-strings is represented by an iarray I_keys of N*( nBits/32 )
elements (e.g. if nBits is 32 , each integer represents 1 bit-string,
if nBits i 64, I_keys1 has two integers for each bit string, etc.).
The returned matrix dimensions are N1 x N2 .
The distance is defined as 1. - similarity , where
The Tanimoto similarity between bitstrings is defined as follows:
The number of the on-bits in-common between two strings divided by the number of
the on-bits in either bit-string.
You can provide a relative weight for each bit in a bit-string as a
rarray R_weights.
In this case the weighted Tanimoto distance is calculated as follows:
distWeighted = 1. - Sum( Wi_of_common_On_Bits ) / Sum( Wi_of_On_Bits )With option
simple the similarity calculation is modified so that
the number of bits in common is divided by the number of bits
in the second bit-string. For example:
Distance({3} {1} 32 simple ) # returns 0.
Distance({1} {3} 32 simple ) # returns 0.5Example:
Distance({1 2 3},{1 2 3},32)
#>M
0. 1. 0.5
1. 0. 0.5
0.5 0.5 0.Instead of the number of bits, one can provide the relative weights for each bit. The dimension of the bit-weight array then becomes the size of the bit-string. The weighted Tanimoto is calculated.
See also:
- Distance(X, X, .., set) to calculate a single distance between two chemical sets
- Score(X, X, .., set)
- Iarray-bits-to-integers{ Iarray({1 0 0 1 1 0 ..} key ) } to generate compressed integer bit vectors
Distance( X_n X_m [[R_Wn R_Wm] r_minScore (0.3) [r_steepness(6.)]] set ) → r_distance [0:1]
retuns a real effective distance between two chemical sets. It is equal to 1 - r_similarity defined by function Score(X1,X2,..set)
See also:
Distance ( M_coor1 M_coor2 ) matrix of distances between the rows of the two input matrices.
Each matrix row may contain any number of coordinates coordinates (3 in 3D space).
For example: Distance(Xyz(a_/1:5/ca) Xyz(a_/10:12/ca) returns a 5 by 3 matrix of distances between Ca-s of the two fragments.
Distance( M_xyz1|as1 M_xyz2|as2 r_dist ) → l_yes_if_closer_than_distyes if any two points or atoms in two sets of coordinates or selections are closer
than the threshold.
if Distance ( as1 as2 r_dist ) then ...is a more efficient version of this condition:
if Nof( Sphere( as1 as2 r_dist )) > 0 Distance ( as [ r_defaultLength=-1.] ) - returns the real array of lengths of tethers for each selected atom or the default value ( -1. ). The default value can be set to any value. Tethers are assumed to be already set, see command set tether. Also note, that the expression
Distance( as_out )
will give the same results if as_out selection was not changed by
another operation; see also special selections.
Example:
|
|
Distance( seq1 seq2 [identity|evolution|new|fast|number|reverse] ) → r
Distance( r_dist_0_to_1|M_dist ) → r|M_Dayhoff_corrected_distances to [0.:10.]
Distance( seqArr[n] seq ) → R_n
Distance( d_0to1|M evolution ) → d|M_Dayhoff_correction_applied
- returns the real measure of difference between two aligned sequences. Zero distance means
100% identity. The distance is calculated by the following two steps:
- d1 = 1.0 - (nResidueIdentities/Min(Length(Seq1), Length(Seq2)) (d1 belongs to [0.,1.] range)
- if there is no
identityoption (or thesequenceoption for a general value to value transformation), the distance is corrected: Distance(Seq1,Seq2) = DayhoffTransformation( d1 )
Transformation practically does not change small distances d1, whereas large distances, especially above 0.9 (10% sequence identity) are increased to take occasional reversals into account. Distances d1 within [0.9,1.0] are transformed to [5.17, 10.] range.
The last function ( Distance( d|M evolution ) allows to apply Dayhoff correction that extends a distance from 0. to 1. to a range 0. to 10. to take into acount the evolutionary time correction (stretching) at larger distances because.
See also:
Distance ( ali ) for distance and seq.identity matrices.
Distance ( alignment ) → M_nxn
Distance( seqArr_n ) → M_nxn
Distance( seqArr_n seqArr_m ) → M_nxm
- returns matrix of pairwise sequence-sequence distances in the alignment.
These distances are calculated with the fast option as follows
- for each pair of sequences in multiple alignments calculate sequence identity according to the alignment as:
the resulting number will be between 0. and 1.1.-(nResidueIdentities)/Min(Length(Seq1), Length(Seq2)) - apply the Dayhoff correction formula that will convert the linear distance to 'evolutionary' distance (kind of evolutionary time needed to arrive at this identity. Where the inifinite time is defined as 10. The resulting distance will be between 0. and 10. and area of 'insignificant linear distances' will be pushed to larger than 1. values. Between two identical sequences D= 0. , while the distance between two 30% different sequences will be around 0.5. The distance goes to an arbitrary number of 10. for completely unrelated sequences.
Example:
read alignment msf s_icmhome+"azurins" # read azurins.msf
NormCoord = Disgeo(Distance(azurins)) # 2D sequence diversity in
#
# calculate pairwise sequence identities
read alignment "aln" name="aln"
n=Nof(aln)
mids = 100*(Matrix(n,n,1.) - Distance(aln )) # the pairwise seq. identities
t = Table( mids, Name(aln), Name(aln) ) # to convert the matrix into pairwise table
t = Table( mids, index ) # a simpler version with i,j Distance ( ali_1 ali_2 [ exact ] )
- returns the real distance between two alignments formed by the same sequences.
The distance is defined as a number of non-gap columns identical between
two alignments.
Two different normalizations are available:
The default normalization is to the shorter alignment. ( Distance ( ali_1 ali_2 ) ). In this case the number of equivalent pairs
is calculated and is divided by the total number of aligned pairs in the shorter
alignment.
This method detects alignment shifts but does not penalize un-alignment of previously aligned residue pairs.
D = (La_min - N_commonPairs)/La_min In the following alignment the residue pairs which are aligned in both alignments
are the same, therefore the distance is 0.
show a1 # La1 = 3
ABC---XYZ
ABCDEF---
show a2 # La2 = 6
ABCXYZ
ABCDEF
Distance(a1,a2) # a1 is a sub-alignment of a2, distance is 0.
0.exact option: normalization to the number of pairs of the longer alignment. By longer we mean the larger number of aligned pairs regardless of alignment length (the latter includes gaps and ends). D = (La_max - N_commonPairs)/La_max Now in the above example, La_max = 6 , while N_commonPairs = 3, the distance is 0.5 (e.g. the alignments are 50% different).
Distance(a1,a2,exact) # returns 0.5 for the above a1 and a2Example showing the influence of gap parameters:
read sequence msf s_icmhome+"azurins.msf"
gapOpen =2.2
a=Align(Azu2_Metj Azup_Alcfa) # the first alignment
gapOpen =1.9 # smaller gap penalty and ..
b=Align(Azu2_Metj Azup_Alcfa) # the alignment changes
show 100*Distance(a b ) # 20% difference
show 100*Distance(a b exact ) # 21.7% difference
show a b
Distance( treeArr i_at separator )
- return the current value of the cluster splitting level set by split command.
Distance( chemarray [pharmacophore] )
- return square matrix of chemical distances. The chemical distance is
defined as the Tanimoto distance between binary fingerprints
Option pharmacophore uses different fingerprints based on ph4-type triplets.
Example:
Distance( Chemical( { "CCC", "CCO"} ) )
Distance( chemarray1 chemarray2 [pharmacophore] )
- return a MxN matrix where M is number of elements in chemarray1 and
N is number of elements in chemarray2 Option pharmacophore uses different fingerprints based on ph4-type triplets.
Example:
Distance( Chemical({ "CCC", "CCO"}) Chemical("CC" ))
Zero distance for non-identical compounds.Sometimes non-identical compounds can give a zero fingerprint distance due to the limitations inherent in finite length fingerprints. To make the distance more representative, one can mix different types of distances, e.g. for two chemical arrays X1 and X2
Mdist = Distance( X1, X2 ) + 0.1*Distance(X1,X2, pharmacophore)
See also: find table find molcart other chemical functions
Eigen ( M ) → X_eigenVectorColums and R_out with eigen values- returns the square
matrix ( n x n ) of eigenvector columns of the input symmetric square matrix
M_ . All n eigenvalues sorted by their values are stored in the R_out rarray.
Eigen value decomposition is be given by three matrices: X, Matrix(3,R_out) and Power(X,-1)
Example of an eigen-value decomposition:
# create a symmetric real matrix which describes a transformation
read matrix name="A" input="""
2. 0.6 0.5
0.6 4. 0.3
0.5 0.3 6.
"""
X = Eigen(A) # calculate eigenvectors...
V = R_out # and save eigenvalues in rarray V
L = Matrix(3,V) # diagonal matrix with eigen values
# note that now A can be reproduced by this calculation : X*L*X^^-1
show A, X*L*Power(X,-1)
# Eigenvectors are X[?,1], X[?,2], X[?,3]
show X[?,1] # 1st eigen-vector Energy ( string ) - returns the
real sum of pre-calculated energy and penalty (i.e. geometrical restraints)
terms specified by the string.
Important: this function does NOT calculate the energy, the terms must be calculated beforehand by invoking one of the following commands where energy is calculated at least once: show energy,
minimize,
ssearch command and
montecarlo command.
Note:
- Allowed terms in the string are
"vw,14,hb,el,to,af,bb,bs,cn,tz,rs,xr,sf"; -
"func"stands for the total of all the terms, both energy and penalty; -
"ener"is only the energy part (i.e."vw,14,hb,3l,to,af,bb,bs,sf" ); -
"pnlt"is only the penalty part (i.e."cn,tz,rs,xr" ). -
load conf
and
load frame
commands fill out all the energy/penalty terms, which are stored in
both stacks and trajectory files (of course the values also depend on a set of
free variables).
You can get the energy/penalty terms of the loaded conformation without explicitly recalculating them using the
Energyfunction, e.g. Energy("func")
Energy ( rs [ simple | base | s_energyTerms ] ) - in contrast to the previous function this function with an explicit residue selection calculates and returns residue energies in an ICM object.
convert the object if is not
of the ICM type.
The energies are calculated according to the current energy terms ,
and also depend on the fixation of the object.
Note: Use unfix only V_//S,V before the function call for standard fixation.
This function can be used to evaluate normalized residue energies for standard amino-acids to detect local problems in a model.
For normalized energies, use the
simple option.
The base option just shifts the energy value to the
mean energy for this residue type.
If the simple or base terms are not used, the
current energy terms are preserved.
The energies calculated with the simple or base option
are calculated with the "vw,14,hb,el,to,en,sf" terms.
The terms are temporarily enforced
as well as the vwMethod = 2 and vwSoftMaxEnergy values, so that
the normalization performed with the simple option is always correct. Do not forget to
build string "ASDF"
unfix only V_//S,V
add column t Name(a_/A full) Energy( a_/A simple ) Energy( a_/A base )
show tThis function will calculate residue energies for all terms and set-ups with the following exceptions:
- electrostatic (
"el") term and electroMethod = "boundary element", "MIMEL", or "generalized Born"
The s_energyTerms argument allows one to refine the energy terms dynamically (see example below).
Example:
read pdb "1crn"
delete a_W
convert
set terms "vw,14,hb,el,to,en,sf"
group table t Energy( a_/A ) "energy" Label(a_/A ) "res"
show t
unfix V_//*
group table tBondsAngles Energy( a_/A "bs,bb" ) "covalent" Label(a_/A ) "res"
show tBondsAnglesSee also: the
calcEnergyStrain macro.
Energy ( conf i_confNumber) - returns the
table of all the energy components for a given stack conformations.
The table has two arrays:
-
sarrayof the energy term names ( .hd ) and -
rarrayof energy values for each energy term ( .ey ) and
Energy ({ stack | conf } ) - returns the
rarray of total energies of stack conformations.
Useful for comparison of spectra from different simulations.
Examples:
read object s_icmhome+"crn.ob"
set terms only "vw,14,hb,el,to" # set energy terms
show energy v_//xi* # calculate energy with only
# side chain torsions unfixed
# energy depends on what variables are fixed since
# interactions inside rigid bodies are not calculated,
# and rigid body structure depends on variables
a = Energy("vw,14") # a is equal to the sum of two terms
electroMethod="MIMEL" # MIMEL electrostatics
set terms only "el,sf" # set energy terms
show energy
print Energy("ener") # total energy
print Energy("sf") # only the surface part of the solvation energy
print Energy("el") # electrostatic energy
print r_out # electrostatic part of the solvation energy
Entropy( R_frequencies ) → r_entropy
Entropy( R_energies r_RT_energy ) → r_entropy
Entropy( seq [simple|R_26aa_prob] ) → r_entropy
returns energy calculated as ∑i pi Log(pi) where pi probabilities are calculated either as normalized R_frequencies or exp( -(E-E_min)/ r_kT_energy ) factors.
If the frequency array contains only one element it is considered as the first probability of an array of two probability that should add up to 1., ie {0.2} is interpreted as {0.2, 0.8}.
The sequence entropy is calculated according to the residue probability from a standard amino acid frequency table. You may substitute it with your own array of 26 numbers. Option simple assumes residue frequencies to be 1/20.
Notes:
- note that the entropy is unit-less generally proportional to the number of comparable high frequency/low_energy states.
- Natural logarith (base e ) is used for the calculation
- for Shannon entropy one needs to multiply the answer by 1./Log(2.) ≃ 1.4427
- if one uses molar energies in kcal/mol , the temperature factor will be RT ≃ 0.6 kcal/mol
Examples:
Entropy({0.5, 0.5}) # 0.693147, two equi-prob. states
Entropy({0.2}) # 0.500402, two states: p=0.2 and p=0.8
Entropy({0.2, 0.2, 0.2}) # 1.09861 three states,
Entropy({10.2, 0.2, 0.2, 0.2}) # 0.275593 one estates dominates
# below the numbers are interpreted as energies, and 1.4 is a temperature factor.
Entropy({-30., -28., -31., -15.}, 1.4) # 0.848429. ΔEs divided by 1.4, exponentials used to calc probs.
Entropy({-30., -28., -31., -15.},100.) # 1.38432 at this high temperature 4 states are almost equi-probable. Error - returns
logical yes if there was an error in a previous command
(not necessarily in the last one).
After this call the internal error flag is reinstalled to no.
The shell error flag can be set to yes with the set error command.
Error ( string ) - returns
string with the last error message.
It also returns integer code of the last error in your script in i_out .
In contrast to the logical Error() function,
here the internal error code is not reinstalled to 0, so that you can
use it in expressions like if( Error ) print Error(string) .
Error ( i_error_or_warning_code ) → l Error ( number ) → s logical yes if an error or warning with the specified code occurred
previously in the script. This call also resets the flag (e.g. Error(415) ).
This is convenient to track down certain warnings or errors in scripts (e.g. detecting
if 'readpdb{read pdb} found certain problems).
Option
number will return a string will previously set error and warning messages.
To clear all bits use the clear error command.
Examples:
read pdb "1mng" # this file contains strange 28-th residue
if (Error) print "These alternative positions will kill me"
read pdb "1abcd" # file does not exist
read pdb "1mok"
clear errorerrorAction , s_skipMessages , l_warn, Warning
Error ( r_x [ reverse ] ) - returns
real complementary error function of real x : erfc(x)=1.-erf(x)) ,
defined as
(2/sqrt(pi)) integral{x to infinity} of exp(-t2) dt
or its inverse function if the option
reverse is specified.
It gives the probability of a normally distributed (with mean 0. and
standard deviation 1./Sqrt(2.)) value to be larger than r_x
or smaller than -r_x.
Examples:
show 1.-Error(Sqrt(0.5)) # P of being inside +-sigma (about 68%)
show Error(2.*Sqrt(0.5)) # P of being outside +- 2 sigma Error ( R_x ) - returns
rarray of erfc(x)=1.-erf(x)) functions for each element of the real array
(see above).
Examples:
x=Rarray(1000 0. 5. )
plot display x Error(x ) {0. 5. 1. 1. 0. 1. 0.1 0.2 }
plot display x Log(Error(x ),10.) {0. 5. 1. 1.}
#NB: can be approximated by a parabola
#to deduce the appr. inverse function.
#Used for the Seq.ID probabilities.
Error( soapMessage )- returns a error string from the SOAP message. (empty string if no error)
This function is used the check the result of calling SOAP method.
See: SOAP services for more details and examples.
[ Exist-pattern | Exist molcart ]
function indicates if an ICM-entity exists or not. Exist ( s_fileName [ write | read | directory ] ) logical yes if the specified file or directory exists, no otherwise.
Options:
writeopen for writingreadopen for readingdirectorythe provided string is a directory (not file)
Exist( collection s_fieldname )Checks if the field exists, e.g.
c = Collection(); c['a']=123
Exist(c,'a') # yes
Exist(c,'b') # no
Exist ( key, s_keyName ) logical yes if the specified keystroke has been previously defined. Examples:
Exist(key, "F1" , Exist( key, "Ctrl-B" )
See also: set key command.
Exist ( object ) logical yes if there is at least one molecular object in the shell, no otherwise.
Exist ( os1 stack ) logical yes if there is a built-in object stack , no otherwise.
Exist ( box ) logical yes if the purple box is displayed, no otherwise.
Exist ( view ) logical yes if the GL - graphics window is activated, no otherwise.
Exist ( gui ) logical yes if the GRAPHICS USER INTERFACE menus is activated, no otherwise.
Exist ( grob display ) logical yes if the grob is displayed.
Exist( connect ) logical yes if the mouse rotations are connected to a graphical object ( grob ) or a molecular object.
Exist( s_table_name sql table ) logical yes if there is an sql table with the specified name exists. It works with the Molcart tables or
tables accessed via the Sql function.
Exist( variable s_varName ) yes if the variable exists in the ICM shell, no otherwise. See also Type( Exist(variable, "aaa") # returns no
aaa=234
Exist(variable, "aaa") # returns yesif (!Exist("/data/pdb/") then
unix mkdir /data/pdb
endif
if(!Exist(key,"Ctrl-B")) set key "Ctrl-B" "l_easyRotate=!l_easyRotate"
if !Exist(gui) gui simple
Exist( chemarray pattern )
returns logical yes if at least one of the elements contains SMARTS search attributes, no - otherwise.
Example:
Exist( Chemical("[C&H1,N]") pattern ) # returns yes
Exist( Chemical("CCO") pattern ) # return no
Exist( s_dbtable sql table )
- returns logical yes if the specified table exists in the database
See also: molcart
Existenv ( s_environmentName ) - returns
logical yes if the specified named environment
variable exists.
Example:
if(Existenv("ICMPDB")) s_pdb=Getenv("ICMPDB")Getenv( ), Putenv( ) .
Extension ( string [ dot ] ) - returns
string which would be the extension if the string is a file name.
Option dot indicates that the dot is excluded from the extension.
Extension ( sarray [ dot ] ) - returns
sarray of extensions.
Option dot indicates that the dot is excluded from the extensions.
Examples:
print Extension("aaa.bbb.dd.eee") # returns ".eee"
show Extension({"aa.bb","122.22"} dot) # returns {"bb","22"}
read sarray "filelist"
if (Extension(filelist[4])==".pdb") read pdb filelist[4] Exp ( real ) - returns the
real exponent.
Exp ( rarray ) - returns
rarray
of exponents of rarray components.
Exp ( matrix ) - returns
matrix
of exponents of matrix elements.
Examples:
print Exp(deltaE/(Boltzmann*temperature)) # probability
print Exp({1. 2.}) # returns { E, E squared }[ Field user ]
function. Field ( s [ s_precedingString] i_fieldNumber [ s_fieldDelimiter] ) - returns the specified field. Parameter
s_fieldDelimiter
defines the separating characters (space and tabs by default). If the field number
is less than zero or more than the actual number of fields in this string,
the function returns an empty string.
The s_fieldDelimiter string
Single character delimiter can be specified directly, e.g.
Field("a b c",3," ") # space
Field("a:b:c",3,":") # colonField("a%b:c",3,"%:") # percent OR colonField("a b c",3," ") # two==multiple spaces in field delim
Field("a%b::::c",3,"%::") # a single percent or multiple colonsMore examples:
s=Field("1 ener glu 1.5.",3) # returns "glu"
show Field("aaa:bbb",2,":") # returns "bbb"
show Field("aaa 12\nbbb 13","bbb",1) # returns "13"
show Field("aaa 12\nbbb 13 14","bbb",2," \n\n") # two spaces and two \n .
# another example
read object s_icmhome+"all"
# energies from the object comments, the 1st field after 'vacuum'
show Rarray(Field(Namex(a_*.),"vacuum",1)) Field ( S , [ s_precedingString] i_fieldNumber [ s_fieldDelimiter] ) - returns an string array of fields selected from S_ string array .
s_fieldDelimiter is the delimiter. If the field number is less than
zero or more than the actual number of fields in this string, an element of the
array will be an empty string.
Examples:
show Field({"a:b","d:e"},2,":") # returns {"b","e"}
s=Field({"aa 2 3.3", "bb 4 1.3", "cc 31a 1.1 3"},2)
# returns {"2","4","31a"}
s=Field({"aa 2 3.3", "bb 4 1.3", "cc 31a 1.1 3"},4)
# returns {"","","3"}Split( ).
Field( as|rs|ms|os [s_fieldName] ) Field( { rs | ms | os } [ i_fieldNumber ] )
Field( os 15 )
returns rarray of user-defined field values of a selection.
Some fields are filled upon reading a pdb file
Atoms. Only one user defined field can be set to atoms, e.g.
read object s_icmhome+"crn.ob"
set field a_//* Random(0.,1.,Nof(a_//*))
show Field( a_//* )
read pdb "1f88" # rhodopsin, many loops missing
Field( a_ 15) # returns 31. residues
Field( a_ "pmid") # iarray[1] with pubmed id, automatically created by read pdb
set field a_/10,14,21 name="pocket"
display cpk Field ( a_/* "pocket" )Residues, molecules and objects.
Three user fields can be defined for each residue and up to 16 for molecules and objects. To extract them specify i_fieldNumber . The level of the selection determines if the values are extracted from residues, molecules or objects. Use the selection level functions
Res Mol and Obj to reset the level if needed. For example: Res(Sphere(gg, a_1. 3.))
selects residues of the 1st object which are closer than 3. A to grob gg .
Upon reading a pdb file the object field 15 contains the number of residues missing from
the ATOM records, but present in SEQRES records due to local disorder.
Example:
read object s_icmhome+"crn.ob"
set field a_/A Random(0.,1.,Nof(a_/A)) number = 2 # set the 2nd field to random values
GRAPHICS.atomRainbow= "yellow/green/blue/blue" # optional redefenition of colors
color a_/* Field( a_/A 2 ) # color by itStandard fields:
- object: "pmid" - integer pubmed id
See also:
- set field as_ [ name= s ] .. ,
- Smooth rs_ to generate 3D-averaged user fields
Selectfunction to select by user defined field (e.g. Select( a_// "x>-1." ) ).
File ( os ) Example:
read pdb "/home/nerd/secret/hiv.ob"
File( a_ )
/home/nerd/secret/hiv.ob File ( s_file_or_dir_Name "length" ) - returns
integer file size or -1.
File ( s_file_or_dir_Name "time" ) - returns
integer modification time or -1. Useful if you want to compare which of two files is newer.
File ( icm_object ) - returns
string file name from which this object has been loaded or empty string.
File ( s_file_or_dir_Name ) - returns
string with the file or directory attributes separated by space.
Note that this function will only work on Unix or Mac, see a`Exist ( s_file .. ) function for cross-platform functions. If file or directory do not exist the function returns "- - - - 0" Otherwise, it contains the following 4 characters separated by space and the file size:
- type character:
- 'f' - regular file
- 'd' - directory
- 'l' - symbolic link
- 'c' - character special file
- 'p' - pipe
- 'r' if you can read the file (or from the directory)
- 'w' if you can write to this file (or directory)
- 'x' if you can execute this file (or cd to this directory)
- file size in bytes
Example:
if File("/opt/icm/icm.rst")=="- - - - 0" print "No such file"
if Field(File("PDB.tab"),2)!= "w" print "can not write"
if ( Indexx( File("/home/bob/icm/") , "d ? w x *" ) ) then
print "It is indeed a directory to which I can write"
endif
# Here the Indexx function matched the pattern.
if ( Integer(Field(File(s_name),5)) < 10 ) return error "File is too small" File ( last ) returns the file name of the last icm-shell script
called by ICM.
In scripts File(last) can be used for the Help section.
See also: Path ( last )
File ( T_IndexTable database ) returns the file name of the first source file indexed. Example:
read index "nci"
File( nci database)
/data/chem/nci.sdf[ Find in array | Find in table | Find chemical ]
function searching all fields (arrays) of a table, and to search patterns in sequences or their names.
Find ( R_source r_value )
Find ( I_source i_value ) - returns index of the source array element which is closest to the value
Example:
Find( {10 20 30 40 50} 43 ) #will return 4 because 40 is the closest value
Find( {1. 2. 3.} 100. ) #will return 3
See also: Index
Find ( table s_searchWords ) - returns
table containing the entries matching all the words given in the
s_searchWords string.
If s_searchWords is "word1 word2" and table contains arrays a and b this "all text search" is equivalent to the expression :
(t.a=="word1" | t.b == "word1") & (t.a=="word2" | t.b == "word2").Examples:
read database "ref.db" # database of references
group table ref $s_out # group created arrays into a table
show Find(ref,"energy profile") & ref.authors == "frishman"
Find ( table s_pattern regexp )
- returns table containing the entries where at least one text column matches s_pattern.
Examples:
add column t { "one" "two" "three" } {"Item1", "Item2" "Item3" }
Find( t "Item[12]" regexp ) # matches first two rows
Find( t "two|three" regexp ) # matches last two rows
[ Find-seq ]
Find( mol_array, array_of_chemical_patterns S_labels )
Find( mol_array, table_with_chemical_patterns )returns a 'sarray of chemical-pattern labels found in the mol_array.
If the table argument is provided as the source of the chemical patterns, the function will look for two columns:
.molarray of chemical patterns- a column called
".LABEL"or".LABELS"in either upper or lower case.
Example:
nProc = 4
Find( chemTable.mol, Chemical( {"c1ccccc1", "[CH3]"} ), {"benzene", "methyl"} )
# or
group table t Chemical( {"c1ccccc1", "[CH3]"} ) "mol" {"benzene", "methyl"} "label"
Find( chemTable.mol, t )
See also: Index chemical, Nof find table , find molcart , nProc
: Find(
returns an
Hint: for rounding numbers you can use Floor( value + 0.5 )
Example:
- returns the
function returning the value for an argument to ICM or an icm-script.
If one runs
e.g.
A summary of the Getarg functions:
If the default value is provided, the returned object is cast to default value's type.
Else the function tries to guess the return type based on the value format.
If the default value is of logical type, the function returns the opposite value
if the argument is found in the list. e.g.
for icm or icm-script arguments like name returns a string with "yes".
For argument name=value returns the argument value converted according
to the default value.
The default value is be returned if the argument is not specified.
Option
returns a concatenated list (`string) of all arguments prepared for interpretation
by a Unix shell. This is convenient for passing arguments further to
a nested script. Trim(Getarg(),all) will return the empty string if no arguments are found.
returns
returns
returns
deletes all arguments and returns the number of them
Testing if the argument exists
returns
returns
returns
Examples :
An example with an icm script:
An example of dialog input:
Another example with a text box
See also
- returns
- returns
- returns
- returns
Example
Example (display plane of the phenyl ring)
returns
In order to be able to access the additional information in the objects' header,
they should be read from PDB using the
Notice that if the object was read with the
Example:
See also
[ Iarray making | Iarray inverse | Iarray bits to integers | Iarray atom numbers | Iarray residue numbers | Iarray stack ]
See also:
- returns
Example:
- returns the image array containing
- returns the image array with textures stored in the
see also
returns array with resized images. By default uses high quality but slow algorithm.
Other algorithms are available by specifying the scaling method:
returns an image with the specified sizes and of the specified color.
May be useful for blending images with a certain color (the default value is 'black').
Example:
returns an image or vectorized (
Example:
See also:
returns InChI International Chemical Identifier.
Example:
See also:
[ Index fork | Index chemical | Index string | Index regexp | Index table selection | Index table label | Index unique elements | Index element in array | Index tree | Index compare | Index atom map ]
See also:
- returns
Options
Example:
Finding indices of duplicate entries
This function can be useful to remove redundant compounds from the set.
Options:
Example:
See also:
returns
finds chemically equivalent projection of atoms for a chemical against itself.
The function returns a
returns index of the currently set tautomer. The tautomers must be pre-built with
Example:
See also
Options:
- returns an integer array of order numbers (indices) of rows with labels equal to
See also:
- returns
Examples:
- this function returns a complement of the input set of indexes. It is similar to a negation of a selection.
Examples:
returns
returns an
- returns a
Example:
- performs element wise comparison of two arrays of the same length and type. Returns a
Example:
X_single_chem2 should be substructure or equal to the X_single_chem1.
- returns
Example:
- returns the
If option
If option
returns
Example
See also:
returns
- calculates the integral
- calculates the integral
This method is now replaced by the setting of the
- returns
Examples:
- returns
Each element of the array may have one of the following values:
See also:
The Laplace operator is a second order differential operator.
The Laplacian of ƒ where f is defined in 3D space as map on a grid is the sum of all the
unmixed second partial derivatives in the Cartesian coordinates xi
- returns
- returns
- returns
- returns
See also:
Example:
creates a linear regression prediction-model like:
Y = 5*A + 10*B + 20The resulting model can then be applied to any table with columns required by the model.
The T_weights table should have two columns:
For example, tables produced by the
A simple model example: Y = 0.7*A + 2.3*B - 10.*C + 5.6
See also:
See also:
returns an array of principal moments of inertia for the selected atoms
in each selected object. The input array can also be a
Example:
- returns the matched substring (or empty string).
Example with parsing swiss id, name and description (see macro readUniprot):
minimal and greedy match Check
Case sensitivity To make the match case insensitive use the
- returns an sarray with all matched expression
- returns an sarray with matched substrings, the resulting array has the same size as the input array
[ Matrix new | Matrix sub | Matrix symmetric | Matrix color | Matrix residue comparison | Matrix table | Matrix tensor | Matrix residue areas | Matrix alignment | Matrix boundary | Matrix stack | Matrix histogram | Matrix grob connectivity ]
- returns square unity
multiples R_row vector nRows times into a matrix. Make sure that nRows is not equal to
a submatrix of specified dimensions. To select only columns or rows,
use zero values, e.g.
function returns a matrix of n rows and 3 columns for each of the
- returns comparison matrix in the specified order.
Example in which we extract cysteine, alanine and arginine comparison values:
Example:
The inverse operation is also possible with the Table ( matrix , S_colNames ) function.
- returns
- returns a
Example:
Returns a matrix n_vertices by n_vertices containing 1. for connected vertices and a large number for unconnected.
Example:
Example:
median-value function.
Examples:
- returns the integer index of the nearest sequence in the alignment.
To get the name of the nearest sequence, use the
Note that there is an obsolete [ Name chemical property | Name soap | Name close sequence | Name string | Name tree | Name chemical | Name conf | Name sequence | Name pred colum | Name object parray | Name image | Name molcart ]
Name of the shell variable
- returns a
Example:
All names of objects in a given class
- returns a string array of object names for the specified class.
Classes:
Subclass of strings: html-objects and scripts
See also:
Example:
- returns
Examples:
- returns
See also:
Example:
- returns
Note that function does not generate a systematic (IUPAC) name. It uses names from the first line
of SD/MOL file. Chemical names can also be set with
See also:
- returns
See also:
- returns
The result of this function can be used for analysis of the prediction model results and can used
together with
Each element in the fingerprint part is SMARTS-like expression (some atom properties used in the prediction model cannot be
expressed as a valid SMARTS expression)
For the default atom properties the output will look like this:
See also
- returns
See also:
- returns
See also:
- returns current database connectionID
- returns
- returns
- returns
- returns
- returns
See also:
[ Namex sequence | Namex image ]
See also:
See also:
Example of a test if a [ Nof tree | Nof chemical | Nof distance | Nof library | Nof molcart | Nof latent | Nof soap ]
- returns
- returns
- returns
See also:
- return an
Options
Examples:
See also:
counts number of records in the
Example:
See also:
See also:
See
returns different norms of a vector, e.g. its Euclidean length, or the size of the range of its values.
Examples:
See also:
The following expression returns 1. for any valid s_type and vector v with non-zero norm:
Examples:
returns
returns
See also:
returns the
Example in which Path finds icm executable runs the script with it:
Versions before 3.5-2 used Path(unix,[
Note, that the current version of ICM stores user preferences in the ~/.config/Molsoft.conf file under Linux.)
- returns a string with simplified separators (useful when you want to compare different paths)
Example:
create an empty parray of the specified cell/element-type.
To store images, use the
Example:
See also:
returns
returns
See description above. Additional functions are listed here.
see above, or applies the model specified as the second argument.
An array of the property names for loaded models can be returned by
the
Examples:
Returns torsion statistical profile predicted by graph Convolutional Neural Network (GCNN) trained on structures
from Crystallography Open Database (COD)
J Chem Inf Model. 2022 Dec 12;62(23):5896-5906. doi: 10.1021/acs.jcim.2c00790. Epub 2022 Dec 1.
Available options:
- adds a name-value pair to the list of ICM arguments. Returns no in case of error.
See
-returns a
- function to push the icm or icm-script arguments (see
See also:
[ Random string ]
Examples:
[ rarray sequence projection | Rarrayinverse | R property transfer via alignment | Rarray properties | RarrayAlignment ]
The values returned with both
To project the conservation onto a 3d chain with its linked sequence in alignment use the
Rarray( R_conserv alignment seq3d) projection function. e.g.
See also:
- returns the swissprot database reference if available.
It is possible to specify the requested field name; the default is "DR".
[ Replace exact | Replace simple | Replace regexp | Chemical replace ]
- returns a
Search a string array and find an element which matches the full s_completeString,
e.g. the
In this case there is not intepretation of the query string. The first occurrence of it
is replaced with the second argument.
Example:
- replace the s_regexp in the source string or array by s_byRegexp using regular expressions.
The latter is a string which may contain back-references.
Example: case-insensitve replacements:
Example:
Note that "(?n)" modifier is needed to make '.' match newline too.
Dehtml-tagging of the html text in a string or a string array:Prep work in html conversion is usually this:
<>
S = Replace( S, " ","\n\n", exact)
S = Replace( S, " "," ", exact)
# finally remove all tags
S = Replace( S, "<.*?>","",regexp)
<>
Example in which we remove href html tags from a column in a table :
Finds a chemical pattern containing one of several Rn groups
and replaces the pattern to the s_smileTO pattern according to the matching
R-groups.
Note that all atoms except the ones connected to the R-groups in s_smartFROM pattern
will only match exactly the same local pattern.
The molecules will be redrawn in 2D after the replacement.
The
Example in which we created a newe table tt with a modified column:
See also:
See also:
- returns the
- returns the
See also
- returns real array of root-mean-square distances between each element of chemarrayand ms_select2.
pharmacophore toggles pharmacophore superposition. ms_select2 - pharmacophore template.
- returns the
See also:
[ Sarray index ]
Example:
[ Score overlap | Score chemset | Score torsion | Score apf | Score model | Score predictions | Score sequence | Score conservation | Score alignment ]
- returns the
This function returns a similarity (0. to 1.) between two sets (arrays) of chemicals.
It is calculated as NAB /( NAA + NBB - NAB ), where N is an effective number is similar compounds calculated as weighted sum of sigmoidly transformed similarities to the power of one half. The original similarity measure is transformed by a sigmoid starting from r_minScore (0.) and ending at 1. The mid point of the sigmoid is at 0.5*(1+ r_minScore ) .
A general form of the sigmoid before it is shifted and squeezed is 1./(1.+exp(-b*t)) where b is r_steepness .
Arguments:
returns free strain(s) for a set of 3D molecules
Calculated as:
where
Probability_Tors[i] = Sum( prof[k]*Exp(-d*d) )
k : 1:36 # 36 bins
d : current_torsion_bin-k # distance in bins between current torsion angle bin and k
The result includes entropy component :
if there are three equal depth minima, even at the bottom of any of them there will be an offset penalty corresponding to entropy
loss associated with locking out two of the three minima.
returns all pairwise APF scores between 3D chemical arrays. Two prerequisities:
Example:
If the two sites (or atoms sets) are not superimposed, use the
siteSuperAPFas1 as2
Categorical or class prediction (e.g. Bayesian classifier). If each data record has a label which can
be either positive or negative (say, 1, or -1) then the success of a prediction method
can be measured by the following measures:
Quality measures to evaluate a regression method predicting numerical values, e.g. Partial Least Squares, or Kernel Regression.
returns an array of scores of sliding no-gap sequences.
these two functions return the match or no-gap-alignment score for one frame or multiple frames with the
The second function template returns a table with the following columns:
See also:
Setup: a multiple sequence alignment, one of the sequences is linked to a structure, you may want to color residues by conservation or other measure of a column in an alignment. For a straight conservation value for each position in an alignment see
See also:
To extract a pairwise alignment of sequences 1 and 2 from a multiple alignment use the
To return a matrix of all pairwise seq. identities, use this:
[ Select break | Select fix | Select neighbors | Select by nmembers | Select graphical | Select expand | Select by atom property | Select_projection | Select_by_text | Select_by atom numbers | Select_patching | Select_lists | Select_by_sequence | Select by alignment | Select by center of mass ]
this function returns pairs of atoms (i) connected with abnormal bond lengths, and, (ii) breaks in the backbone of
a polypeptide (even if a 'C' carbon and the following 'N' are not bound covalently).
Example in which we find residues flanking the missing loop:
- returns a sub-selection of as with atoms having the specified number of covalent neighbors.
Example:
See also:
These functions allow one to select objects according to the number of molecules in them, and molecules according to the number of residues in them.
Allowed comparison operations : ==, >, >=, <, <=, != .
Example:
- returns residue selection of the residue selection strings which can be returned with the
This function by default (or with option
For example, if you have a residue selection, e.g. a_/1,2,5,6
Option
Interacting atoms.selecting atoms interacting with the source atoms according to a particular energy term.
It is required that the source atoms are in the current ICM object and
Tether destination atoms.In case of
- returns molecular selection of all chains with sequences similar to seq .
Arguments and options:
To select the closest residue from a center of mass of one selected residue, use the
E.g.
To find the closest residue to residue 44 in the above example, use the table approach, e.g.
A faster implementation of the same task with the [ Dna to rna conversion | Reverse complement | Sequence array ]
Examples:
Example:
See also:
randomly change order of elements of an array or a sequence of characters.
Example:
- returns a compact binary representation of the entire graphical view (also known in ICM as a
The data is packed into a single-element
To display the view use the display parray_slide command, e.g.
- returns a
See also:
- returns a
The
Example:
[ Smooth | Smooth matrix | Smoothrs | Smooth alignment | Smooth map ]
The values in the source matrix get transformed according to a Gaussian 2D transformation
in which the values
Examples:
returns an array of real roots of a quadratic equation ax2 + bx + c = 0
By default only real roots are found.
Option
See also:
returns an array of real roots of a qubic equation ax3 + bx2 + cx +d = 0
By default only real roots are found.
Option
Example:
See also:
this function returns a
The atoms will be searched in the specified selection as_whereToSelect
if the second selection is explicitly specified.
If only one atom selection is specified,
the atoms will be selected from the same object.
The selection level functions (
For example, selection
Adjusting for the van der Waals radii, vdW gap. Use negative distance values to indicate a different mode of the Sphere function.
Sphere can also correct for the van der Waals radii if you specify the negative radius.
Values < -1. indicate vdW gap ( -1.15 means 15% larger than the sum of vdW radii).
In this case it is interpreted as a ratio of the inter-atomic distance to the sum of van der Waals radii.
For example,
The negative sign just flags the program to use the distance to vwlimit ratio instead of the distance.
The value of -1.15 roughly corresponds to 3.5 .
Example:
this function returns a
Example:
See also:
A SOAP message is special XML text which contains :
The following example form a SOAP request to the google search service.
See
- returns the sorted array. Option
[ Split tree | Split regexp | Split multisep | Split chemical ]
# "" to split into characters
Multiple spaces are treated as one space, while all other multiple separators
lead to empty fields between them. If s_Separator is an empty string
(""), the line will be split into individual characters. To split a multi-line
string into individual lines, use Split( s_, "\n" ).
Returns
Example:
- returns an
Useful separators:
Examples:
- takes a
where name and value can be any text which does not contain sep1 or sep2
returns a
Example:
See also: Sum-multisep
See also:
Summary:
Chemical match
- returns the
Option
See also:
Different kinds of atom equivalences
Two version of the
Optimal path
This function returns the number of atom pairs (or selftethers) used in the calculation in
Matrix of static RMSDs
returns the "optimal path" matrix of srmsd values where for each two molecules the smallest srmsd is accumulated.
The dimensions of the matrix are
Matrix of superposition errors
In this case the selections are split into molecules (just as in the Srmsd ( as1 as2
If option
Pharmacophore distance
- returns
Example:
See also:
- returns
[ String substring | String date | String mol | Alignment_as_text | Ali_seq_project | Seq_ali_project | String alternative | String selection | String slide gui | Chem formula ]
Detailed descriptions:
the format specifications are described in the
See also :
generates a string buffer in
generates string buffer in
Example:
This function is equivalent to the question mark operator in C, e.g. condition?choice1:choice2
Example:
converts a selection into a compact string form.
Continuous blocks of selected elements in different molecules or objects are separated
by vertical bar ( | ) which means logical or ( e.g.
Options:
retrieves the string with the window layout information which is stored in the slide.
Example:
See also:
Examples:
[ Sum chemical | Sum image ]
- returns
Empty values are skipped. This function can be used to 'shrink' sparse tables
Example:
See also:
Merges elements of
This function can be used to merge several PDB ligands into one molecule
Example:
See also:
creates an image array consisting of blended images from arrays imageArray_a and imageArray_b.Arrays should have the same size and each image pair in the arrays should have matching sizes as well.
The r_bweight parameter specifies how much of the color in resulting image should be taken
from the second array images. r_bweight should be a value between 0 and 1.
See also:
See also, [ Table url_decoder | Alignment as table | Residue correspondences | Table matrix | Table pairs | Table stack | Table plot | Table model | Table model chem | Table distance | Protonation states table ]
- returns a table of icm residues from the
- returns the
This table may look like this:
If an alignment is linked to a 3D molecule, all cell of this row will show
both sequence numbers, as well as residue numbers of the linked 3D molecule, see
example below. The columns names are composed of letter 'p' for position and alignment position (eg
Note that in contrast to the previous function, this function looks like an alignment
and has the same orientation. Each row corresponds to a different sequence,
the sequence name is stored in the first column, while other columns contain
residue numbers in the selected alignment positions.
Example:
- returns
The inverse operation can be done with Matrix ( table , S_colNames ) function.
This function will return a table
with three or five columns, named
Example:
takes the svg output of the
Example:
returns a table with three columns:
Example:
Returns table with the following columns
inverse option returns fragments which are not present in the model. ~w and ~wRel columns are omitted in this case.
Example:
- takes a
Example in which we find the shortest hydrogen bond in crambin:
See also:
- returns the
- returns the
convert to integer values or arrays.
Example in which we form two classes for positive and negative values.
Useful, e. g. in
A more general splitter:
- these functions recode source, replacing each value found in labels array
by the respective value from the values array. Thus, values array should have
the same number of elements as the labels array. Alternatively, it may contain an extra element,
and that last element will be interpreted as the default value for everything from the source not listed in labels.
Example:
See also:
convert to real values or arrays.
Example:
Support for special values in real arrays.Section
See also:
convert to integer values or arrays.
See also:
[ Dna translate ]
returns one or n transformations in the form of
one 12*n long vector. Here os_1 means selection of one single object (e.g.
All columns in the result table will be assigned the same type which is determined from column types of the source table.
The result type can be either
Optional argument i_nameColumn specifies the column number in the source table which will be excluded from the transposition
and it's values will be used to assign column names in the result table.
Example:
- returns
- returns
- returns
iteratively identifies the smarts patterns and deletes it.
Arguments:
See also:
This function finds the matching regular expressions in the source sequence
and deletes it.
Note that the order is important and the longer patterns need to precede the shorter ones.
The pattern can be N-terminal (use ^) , a fragment in the middle, or C-terminal (use dollar $ )
There is a built in shell array called
[ Type soap | Type molcart ]
- returns a
The non-ICM types can be changed with the
The molecule type can be reset with the set type ms_ s_type command,
e.g. (
- this function will always return a sarray with types for each selected unit. With
See
- returns
See also:
- returns the list of sorted unique values or their number of occurrences ( the
Example:
See also:
See also:
Extracts a content from the SOAP message ( See
- returns either basic type (for strings integers or reals ) or SOAP
object which may consists of these basic types grouped together into arrays or structures
See [ Vectorproduct | Vectorsymmetrytransformation | LatentVector ]
By the way the vector dot product is just
See also:
See also:
- returns
Option
- returns
- returns either
See detailed descriptions below
volume of a 3D box defined by coordinates of two opposing corners (see also the
Here are the components of the View vector:
Example on how to translate a defined physical coordinate to the front clipping plane or a center:
To rotate an your view around a screen X,Y, or Z axis by an angle use
See also
set view and
set view.
Info(display)
rotateView macro
- returns 37-
[ Xyz points | Xyz rings | Xyz mesh | Xyz fract | Xyz transformed xyz | Xyz chemical match | Xyz vector2matrix | Xyz axes ]
- returns
Example:
<>
read pdb "2l8h"
display a_N xstick
g_ring_centers = Grob( Xyz( a_N//RA ring ) dot ) # centers or aromatic rings
display g_ring_centers
<>
Transforming absolute coordinates to fractional (i.e. the unit cell) coordinates and
the inverse transformation.
The cell parameters can be taken from an object or from the 3 or 6 cell parameters.
Example:
returns a new Nx3 coordinate matrix with the source coordinates transformed according to
R_12abs_transformation or i_symm_transformation of the specified symmetry group.
If i_symm_transformation is greater than the number of symmetry operations in the specified
symmetry group, the transformation goes to the 26 neighboring crystallographic cells.
The central cell can be determined in three different ways:
If option
Translation to the vicinity of automatically or statically defined center (option
Also see the
returns three row-vectors A, B and C corresponding to the R_6cell parameters.
The same vectors can be obtained as columns of the sarray of sequence names in which the sequence matched the pattern, e.g.
make sequence 10 # generates 10 random sequences
Find( "*A?[YH]*" sequence )
Floor ( r_real [ r_base ] )
- returns the largest
real
multiple of r_base not exceeding r_real.
Floor ( R_real [ r_base] )
- returns the
rarray
of the largest multiples of r_base not exceeding components of the
input array R_real.
Default r_base= 1.0 .
rr = { 0.1, 0.5, 1.2, 1.7 }
Floor( rr + 0.5 )
See also: Ceil( ).
Formula( chemarray )sarray of compounds' molecular formulas.
icm directly, specify arguments after the -a option,
icm -s -a t=2 verbose c='some text' # three arguments passed to icm
icm_script t=2 verbose c='some text' # three arguments passed to icm_script
Examples:
if Getarg(help) quit HELP
mid = Getarg( "-mid",no,delete) # logical files = Getarg(list,delete) # all args without '-'
files = Getarg(input,delete) # file names (undashed args), appended with 'keep' and stdin if necessary
outfiles = Getarg(output,delete) # file names
files = Getarg(mol,delete) # same for .sdf* files
c=Collection()
c["a" ] = Getarg("-a",test) # logical to activate the option
c["a_params" ] = Getarg("-a","10:30",delete) # defaults and params
c["m"]=Getarg("-m",test); c["mfrto"] = Getarg("-m","100:500",delete) Getarg ( s_icmargName [s_default] [ delete ] ) Getarg ( s_int_argName [i_default] [ delete ] ) Getarg ( s_real_argName [r_default] [ delete ] ) Getarg ( s_log_argName [l_default] [ delete ] )Getarg("-x",yes) will return
no if the option was specified).
delete extracts the variable from the list.
Getarg( ) Getarg( list|keep [delete] )sarray of non-option arguments (usually they are file names). Option keep adds the "stdin" for dash or no arguments, and adds keyword keepto keep the file open for multiple 'chunk' access to it.
Getarg( name )sarray of argument names
Getarg( set )sarray of argument values
Getarg( delete ) Getarg( s_argName [find|test] )yes if the argument can be found in the list in any form.
Getarg( s_argName [name] )yes if the argument is in the list as the name only (rather than the
name=value pair). E.g. -verbose will return yes, and -verbose=2.3
will return no.
Getarg ( i_pos gui )string which contain a user input after GUI dialog execution using Askg function.
The example above may be called from the shell like:
if Getarg("-L" find) print "-L was found"
t = Getarg("time","1.",delete)
s = Getarg("sequence","ABC")
Getarg("-L" yes ) # returns no in this case, yes is the default
Getarg("-L" no ) # returns yes since no was the default
args = Getarg(name)
wrongArgs = NotInList({"s","t"} ,args)
if wrongArgs print " error> illegal arguments ", Sum(wrongArgs)> icm -a time=1.5 sequence="ADEGFKL" -L file1 file2> cat script.icm
#!icm -s
x=Getarg("x","3")
y=Getarg("y","a b c")
show x,y
> script.icm x=33 y="d e"
33, "d e"buf = "#dialog{\"Select InSilco Models\"}\n"
buf += "#1 s_Some_Input (some text)\n"
buf += "#2 l_Check (no)\n"
buf += "#3 i_Number (4)\n"
Askg( buf ) # run the dialog
print Getarg( 1 gui ) Getarg( 2 gui ) Getarg( 3 gui )txw_ spec :
#dialog{ "Sample Dialog" }
# txw_Enter_Text ()
txt = %s_out # s_out is not a safe place (might be overwritten)
print Length(txt)Putarg , Getenv, script .
Getenv ( s_environmentName [s_default] )
- returns a string of the value of the named environment variable.
If the default string is provided, it is used if the variable is not found.
Example:
See also:
user = Getenv("USER") # extract user's name from the environment
if (user=="vogt") print "Hi, Gerhard"
Getenv("HOME","you are homeless :-(") # use default if HOME is not found
/home/ruben/
Getenv("HOME_MISSPELLED","you are homeless :-(")
you are homeless :-(
Getenv("HOME_MISSPELLED") # errorExistenv( ), Putenv( ) .
Gradient( )
- returns the real value of the root-mean-square gradient
over free internal variables.
Gradient ( vs_var )
- returns the rarray
of pre-calculated energy derivatives with respect to specified variables.
Gradient ( as | rs )
- returns the rarray of
pre-calculated energy derivatives with respect to atom positions
(G[i] = Sqrt(Gxi*Gxi+Gyi*Gyi+Gzi*Gzi))
The function returns atom-gradients for atom selection ( as_ ) or
average gradient per selected residue, if residue selection is specified ( rs_ ).
You can display the actual vectors/"forces" (-Gxi, -Gyi, -Gzi) by the
display gradient
command.
Important: to use the function, the gradient must be pre-calculated
by one of the following commands:
show energy,
show gradient,
minimize .
Example:
read object s_icmhome+"crn.ob"
show energy # to calculate the gradient and its components
if (Gradient( ) > 10.) minimize
show Max(Gradient(a_//c*) # show maximum "force" applied to the carbon atoms
Grob( M_NxM )grob for 3D surface function: X=i,Y=j,Z=~~M_NxM
Grob( M_Nx3_xyz dot )grob of dots with xyz from M_Nx3_xyz Grob( M_Nx3_xyz r_ra ball )grob of spheres with centers from M_Nx3_xyzgrob of dots with xyz from M_Nx3_xyz and radius r_ra Grob ( "arrow", { R_3 | R_6 } )
- returns grob containing 3D wire arrow between either 0.,0.,0. and R_3, or
between R_6[1:3] and R_6[4:6].
Grob ( "ARROW", { R_3 | R_6 } )
- returns
grob
containing 3D solid arrow. You may specify the number of faces
by adding integer to the string: e.g. "ARROW15" (rugged arrow) or
"ARROW200" (smooth arrow).
See also:
GROB.relArrowSize.
Examples:
GROB.relArrowSize = 0.1
g_arr = Grob("arrow",Box( )) # return arrow between corners of displayed box
display g_arr red # display the arrow
g_arr1 = Grob("ARROW100",{1. 1. 1.})
display g_arr1
Grob ( "cell", { R_3 | R_6 } )
- returns
grob
containing a wire parallelepiped for a given cell.
If only R_3 is given, angles 90.,90.,90. are implied.
Grob ( "CELL", { R_3 | R_6 } )
- returns grob
containing a solid parallelepiped for a given cell.
If only R_3 is given, angles 90.,90.,90. are implied.
Example:
read csd "qfuran"
gcell = Grob("CELL",Cell( ) ) # solid cell
display a_//* gcell transparent # fancy stuff
Grob ( "distance", as_1 [ as_2 ] )
- returns grob with the distance lines. This grob can be displayed
with distance labels (controlled with the GRAPHICS.displayLineLabels parameter).
With one selection it returns all possible interatomic distances within this
selection. If two selections are provided, the distances between the atoms of
the two sets are returned.
Example:
build string "se ala his trp"
g = Grob( "distance", a_/1/ca a_/2/ca )
display g
GRAPHICS.displayLineLabels = no
display new
Grob ( "label", R_3, s_string )
- returns grob containing a point at R_3 and a string label.
Grob ( "line", R_3N )
- returns grob
containing a polyline R_3N[1:3], R_3N[4:6], ...
Example:
display a_crn.//ca,c,n
g = Grob("line",{0.,0.,0.,5.,5.,5.}) # a simple line (just as an example)
display g yellow
gCa = Grob("line",Rarray(Xyz(a_//ca))) # connect Cas with lines
display gCa pink # display the grobs
Grob ( "SPHERE", r_radius i_tesselationNum )
- returns grob containing a solid sphere. The i_tesselationNum parameter may be 1,2,3..
(do not go too high).
Example:
display a_crn.//ca,c,n
# make grob and translate to a_/5/ca
# Sum converts Matrix 1x3 into a vector
g=Grob("SPHERE",5.,2)+Sum(Xyz(a_/5/ca))
# mark it with dblLeftClick and
# play with Alt-X, Alt-Q and Alt-W
display g red
- returns Grob ( "TORUS", r_radius r_radius2 [R_normalVector] [i_quality] ) grob containing a solid torus.
- returns Grob ( "ELLIPSOID", r_radius r_radius2 [R_normalVector] [i_quality] ) grob containing a solid ellipsoid.
- returns Grob ( "CYLINDER", r_radius r_height [R_normalVector] [i_quality] ) grob containing a solid cylinder.
t = Grob("TORUS", 1.2 0.2 )
e = Grob("ELLIPSOID" 1 0.4 )
display smooth t red
display smooth e bluebuild smiles "(CC(C)Cc1ccc(cc1)C(C)C([O-])=O)"
display xstick a_
find chemical a_ "c1ccccc1" # result is stored into as_out
n = Normalize(Vector( Rarray(Xyz(as_out[2])-Xyz(as_out[1])) Rarray(Xyz(as_out[3])-Xyz(as_out[1])) ),"euclidean" ) # normal
gr_plane = Grob( "CYLINDER", 2. 0.05, n ) + Mean( Xyz( as_out ))
display smooth transparent gr_plane
Grob( grob R_6rgbLimits )
returns a grob containing selection of vertices of the source grob.
The vertices with colors between the RGB values provided in the
6-dim. array of limits will be selected. The array of limits consists
of real numbers between 0. and 1. :
{ from_R, to_R, from_G, to_G, from_B, to_B }
If you want a limit to be outside possible
rgb values, use negative numbers of numbers larger than 1., e.g.
a selection for the red color could be: {0.9,1.,-0.1,0.1,-0.1,0.1}
The grob created by this operation has a limited use and will contain only vertices
(no edges or triangles).
This form of the Grob function can be used to find out which atoms or residues are
located to spots of certain color using the Sphere( grob as_ ) function.
Example:
build string IcmSequence("ADERD") # a peptide
dsRebel a_ no no
g=Grob(g_electro_def_ {0.9,1.,-0.1,0.1,-0.1,0.1} ) # red color
display g_electro_def_ transparent
display g
show Res(Sphere( g, a_//* 1.5))
See also: color grob by atom selection, and GROB.atomSphereRadius .
Group ( R_n_atoms as_n_atoms "min"|"max"|"avg"|"rms"|"sum"|"first" ) → R_resArray Group ( I_n_atoms as_n_atoms "min"|"max"|"avg"|"sum"|"first" ) → I_resArray Group ( as_atomSelection "count" ) → I_resArrayOfNat
returns an array of atoms properties aggregated to a per-residue array.
One of the following functions can be applied to the atomic values:
The function name is case-insensitive (you may use "Min" or "MIN").
Example:
read pdb "1crn"
show Group( a_A//* "count" ) # numbers of atoms in residues
show Group( Mass( a_A//* ) , a_A//* "sum" ) # residue masses
show Group( Mass( a_A//* ) , a_A//* "rms" ) # residue mass rmsd
Header ( os )sarray with the PDB entry information stored in the requested objects.
PDB entry information is stored in objects in HTML format. Use Header( os1_ )[1] for a single string.
read pdb command with the header option.
read pdb html option the header will be in html format,
while it if the header option was used instead, the entire header will be stored as is.
read pdb "1crn" html
h1 = Header( a_1crn. )[1]
set property h1 htmlread pdb .
matrix [ n,2], where n is number of cells, the first row contains a number
of elements in each cell and the second row contains mid-points of each cell.
Histogram (I_inputArray )
- returns matrix with a histogram of the input array.
Histogram ( R_inputArray, i_numberOfCells [, R_weights ] )
- returns histogram matrix [ i_numberOfCells,2] in which the whole range of the R_inputArray
array is equally divided in i_numberOfCells windows. An array of point weights can be provided.
Histogram ( R_inputArray, r_cellSize)
- returns matrix [ n ,2], dividing the whole range of R_inputArray equally into r_cellSize windows.
Histogram ( R_inputArray, r_from, r_to, r_cellSize )
- returns matrix
[ n,2], dividing into equal cells of r_cellSize
between minimum value, maximum value.
Histogram ( R_inputArray, R_cellRuler [, R_weights ] )
- returns matrix [ n,2],
dividing the range of the input array according to the R_cellRuler
array, which must be monotonous. An array of R_weights of the same
size as the input array can be provided.
Examples:
plot display Histogram({ -2, -2, 3, 10, 3, 4, -2, 7, 5, 7, 5}) BAR
a=Random(0. 100. 10000)
u=Histogram(a 50)
s_legend={"Histogram at linear sampling curve" "Random value" "N"}
plot display regression BAR u s_legend
a=Random(0. 100. 10000)
b=.04*(Count(1 50)*Count(1 50))
u=Histogram(a b)
s_legend={"Histogram at square sampling curve" "Random value" "N"}
plot display BAR u s_legend
b=Sqrt(100.*Count(1 100))
s_legend={"Histogram at square root sampling curve" "Random value" "N"}
plot display green BAR Histogram(a b) s_legend
Iarray( [i_n=0 [i_default=0]] ) Iarray( R|S|I ) → I Iarray( I reverse ) → I_reverseOrder Iarray( I key ) → I_compress01intoInts # obsolete Iarray( stack ) → I_nofVisits Iarray( as ) → I_atomCodes Iarray( as topology ) → I_atomSymmetryNumbers Iarray( rs|ms|os ) → I_nAtomsInEachRes|Mol|Obj
Iarray ( i_NumberOfElements [ i_value ] )
- returns iarray of i_NumberOfElements elements set to i_value or zero.
You can also create an zero-size integer array: Iarray(0) .
Iarray ( rarray )
- returns iarray of integers nearest to real array elements in the direction
of the prevailing rounding mode magnitude of the real argument.
- converts sarray into an Iarray ( sarray ) iarray.
Examples:
a=Iarray(5) # returns {0 0 0 0 0}
a=Iarray(5,3) # returns {3 3 3 3 3}
b=Iarray({2.1, -4.3, 3.6}) # returns {2, -4, 4}
c=Iarray({"2", "-4.3", "3.6"}) # returns {2, -5, 3}
Iarray ( iarray reverse )
- converts input real array into an iarray with the reversed order of elements.
Example:
Iarray({1 2 3} reverse) # returns {3 2 1}
See also: Sarray( S_ reverse ), Rarray( S_ reverse ), String(0,1,s)
- returns a shorter vector of integers if Iarray( I_nBitVector key ) n/32 elements, in which every 32 array values
of zeros and non-zeroes are compressed into one integer. The number of elements n does not need
to be a multiple of 32, the missing elements will be assumed to be zero.
Example:
Iarray({1 0 1 0 0 0 0},key) # returns {5}
Iarray({1 1 1 0 0 0 0},key) # returns {7}
key ) → M_nxm_Tanimotos
iarray of relative atom numbers in a single object.
This iarray can be saved and later reapplied with the Select ( os_ I )
function. If you selection covers more than one object, the function returns an error.
Example:
build string "se ala"
ii = Iarray( a_//c* ) # returns {6,8,12}
Select( a_ ii ) # returns three carbonsiarray of residue numbers for an input selection.
build string "ala glu"
Iarray( a_/ number ) # residue level
Iarray( a_// number ) # atom level
- returns the Iarray( stack ) iarray of the numbers of visits for each stack conformation.
This is the same number as shown by the nvis> line of the show stack command.
Example:
show stack
iconf> 1 2 3 4 5
ener> -15.3 -15.1 -14.9 -14.8 -13.3
rmsd> 84.5 75.3 6.4 37.2 120.8
naft> 3 0 4 0 2
nvis> 10 9 8 1 4
Integer(stack) # returns { 10 9 8 1 4 }
icm.se -file format.
IcmSequence ( { sequence | string | rs }, [ s_N-Term, s_C-Term ] )
- returns multiline string with full (3-char.) residue names which may be a content of an icm.se file.
The source of the sequence may be one of the following:
IcmSequence(1crn_m)
"ASDGFRE", or "SfGDA;WER" .
rs_ , (e.g. a_2,3/* ).
Rules for one-letter coding:
AaA for ala Dala ala )
; ) ( e.g. AAA;WWW )
If the source of the icm-sequence is a 3D object, the proline ring puckering is analyzed and
residue name prou is returned for the up-prolines (the default is pro ).
The N-terminal and C-terminal groups will be added if
their names are explicitly specified or an oxt atom is present in the last residue
of a chain. Here are the possibilities for automated recognition of C-terminal residue:
IcmSequence( a_/* ) # C-terminal residue "cooh" will be added if oxt is found
IcmSequence( a_/* "","" ) # no terminal groups will be added
IcmSequence( a_/* "","@coo-" ) # "coo-" will be added only if oxt is found
IcmSequence( a_/* "nh3+","coo-" ) # "nh3+" and "coo-" will always be added
The resulting string can be saved to a ICM mol-sequence file and further edited
for unusual amino-acids (see icm.res ).
Examples:
In the last command the ampersend means that the C-terminal residue will only be
added if an write IcmSequence(seq1) "seq1.se" # create a sequence
# file for build command
show IcmSequence("FAaSVMRES","nh3+","coo-") # one peptide with Dala
show IcmSequence("FAAS.VMRES","nter","cooh") # two peptides
show IcmSequence("AA;MRES","nter","cooh") # two peptides
read pdb "2ins"
write IcmSequence(a_b,c/* ,"nter","@cooh") "b.se" # .se file for b
# and c chainsoxt atom is present in the last residue.
There is a build string command to create a single or multiple chain
peptides. Example:
build string "SDSRAARESW;KPLKPHYATV" # two 10-res. peptides
See also icm.se for a detailed description of the ICM-sequence file format.
Image( slides ) slide thumbnails. E.g.
group table t Image( slideshow.slides ) Image( grob texture )grob
set texture
Image( images i_newWidth i_newHeight [s_method] )
method description
"fast" the fastest method, has the lowest quality of scaling
"bilinear" usually about 1.5-2x times slower than "fast" but produces images of higher quality
"glu" driver-dependent, requires the graphics to be enabled, may have limitations on evenness of requested dimensions.
The quality is usually high, the speed depends on driver implementation.
Image( i_width i_height [s_color ("black")] )I = Sum( Image(100,60,"red"), Image(100,60,"#00FF00") ) Image( X_chemarray [i_width (300) [i_height (300)]] [s_displayOptions] [ vector ] )svg ) array with 2D depictions of chemicals. Optionally you can specify width, height and display options
Display options string is described in write image chemical . Option vector generates vectorized .svg array of strings, instead of an pixelized
add column t Chemical( {"CC1CC1","CCO", "CCCC1=NN(C)C2=C1N=C(NC2=O)C1=CC(=CC=C1OCC)S(=O)(=O)N1CCN(C)CC1"} )
S_svgs = Image( t.mol vector )image parray, Sum image, Color image write image chemical
InChi( X_chem [key] ) → S_InChi_or_InChiKeyInChi(Chemical("CC(C)Cc1ccc(cc1)C(C)C(O)=O"))
add column t InChi(t.mol key) name="InChiKey"Chemical
Index( s|seq s|seq_sub [i_skip|last] ) → i_posSubInStr Index( s|S_source[n]> <s|S_pattern[n]> exact|simple|regexp [<i_start] ) → |I_pos[n] # length for regexp match is in i_out|I_out Index( S s regexp all ) → I_matchedPos Index( Sn s {regexp|exact} ) → In_indexInEachElement Index( ali seq ) → i_posSeqInAli Index( ali {rs | selection column} ) → I_columnsInAlignment Index( ali|table|tree selection ) → I_selectedEntries Index( fork [system|all] ) → i_proc|pid|nof_children Index ( {I|R|S}, {I|R|S} compare ) → C_["A","AB","B","BA"] Index( I|R|S i|r|s ) → i_1st_matching_pos Index( I|R|S i|r|s all ) → I_matchedIndexes Index( I|S unique ) → I_uniqueValueIndexes Index( Im_indexes n_max inverse ) → I_n-m_complementaryIndexes Index( map [cell] ) → I_mapLimits | I_xrCellLimits (see Map(m I_xrCellLimits) Index( object ) → i_currObj Index( rs ) → I Index( site seq i_no ) → I_fromToList Index( slideArray s_name ) → i_slideByName ( 0 if not found) Index( T ) → I Index( T i_label label ) → I_labeledEntries (colored in GUI) Index( T sql problem ) → I_rejectedRowIndexes Index( tree center [r_threshold] ) → I_centers Index( S_smi smiles problem ) → I_illegal_smiles Index( Xm s_smi [select] ) → I_matches_in_Xm Index( Xm Xk_query [sstructure|r_taminoto] ) → I_n_matches_in_Xm (n<=m) Index( X1a X1b_sub atom map ) → I_matching_atoms_in_X1a Index( X i_RgroupNumber [group] ) → I_ringNumber
the current of process number filled by the Index( fork ) fork command. This number is zero for the parent process.
process ID of the spawned child (is nonzero in children, zero in parent).
Index( fork system )
number of spawned children (is zero in children, nonzero in parent).
Index( fork all ) fork , wait Nof( fork ) the number of available cores in a computer
Index( chem_array , chemical_or_chemarray , [ sstructure [group] ] [ stereo ] [ salt ] )iarray of indices of compounds from the first chem_array
that contain any of identical compounds (the default), or substructure patterns (the sstructure option) from the chemical array.
Example in which we find the nitro compounds among the known drugs:
read table "oralDrugs.sdf" name = "drugs"
modify oralDrugs.mol delete salt
modify oralDrugs.mol delete salt simple # leaves only the 1st molecule
nitrocomps= t[Index(t.mol Chemical("N(=O)O") sstructure )]
group option toggles a special search mode when all atoms in the pattern except the attachment
points require the exact chemical match.
stereo option chirality will be taken into account.
salt option all molecule fragments will be matched. (useful for exact mode)
add column t Chemical( {"CCC","CCO", "CC", "CCC", "CCC.C", "CC", "CCC"} )
Index( t.mol t.mol[1] ) # find all occurrences of the first molecule ( disregarding stereo and salt )
Index( t.mol t.mol[1] salt ) # find all occurrences of the first molecule (salt is taken into account )
Index( {"C1CC2","CCO", "CxC", "CCC", "CCC.C", "CC", "CCC"} smiles problem )
1
3 Index( chem_array , chemical_or_chemarray r_distThreshold )returns less than r_distThreshold to at least one compound from chemical_or_chemarray.iarray of indices of compounds from the first chem_array with chemical distance Index( chem_array exact [ salt ] [ stereo ] )returns iarray of indices of duplicates (entries found more than once).
Index( X exact ) : selects duplicates by only the main molecule, considers different salts and stereoisomers identical
Index( X exact salt ) : selects duplicates by considering all molecules, so different salts appear as non-duplicate
Index( X exact stereo ) : selects duplicates, different stereo isomers are taken into account
Index( X exact salt stereo ) : selects duplicates by considering stereo isomers and considering all molecules
add column t Chemical( {"CCC","CCO", "CC", "CCC", "CCC.C", "CC", "CCC"} )
show Index( t.mol exact ) # returns {4,5,7,6}
delete t[ Index( t.mol exact salt ) ] # remove duplicates. preserves different saltsNof Find find table find molcart
Index( S_smiles , smiles problem ) → I_indeces_of_illegal_smiles_stringsiarray of incorrect smiles strings. Example
Index( {"C1CC2","CCO", "CxC", "CCC", "CCC.C", "CC", "CCC"} smiles problem )
1
3 Index( as enumerate ) → collection_of_equivalent_self_atom_mappingsparray which contains one or several iarray(s) arranged into an ivector( ivector is a subtype of parray) .
Example:
build smiles "C1=CC=CC=C1"
x = Index(a_//!h* enumerate)
Nof(x) # returns 12 possible iarray mappings (six shifts for flipped and non-flipped)
show x # all mappings
1,2,3,4,5,6
1,6,5,4,3,2
2,1,6,5,4,3
2,3,4,5,6,1
3,2,1,6,5,4
3,4,5,6,1,2
5,4,3,2,1,6
5,6,1,2,3,4
4,3,2,1,6,5
4,5,6,1,2,3
6,1,2,3,4,5
6,5,4,3,2,1
map1 = x[1] # creates an integer array Index( as tautomer ) → index_of_current_tautomer_from_mask_hydrogenbuild tautomer command.
The tautomer can be set either by set tautomer command or explicitly by masking hydrogen with set command.
build smiles "N(C(N=C1N)=O)C=C1"
build tautomer a_1
set tautomer a_1 3
Index( a_1 tautomer )build tautomer, set tautomer commands
Index ( { s_source | seq_source }, { s_pattern | seq_pattern }, [ { last | i_skipToPos ] )
- returns integer value indicating the position of the pattern substring in the source string,
or 0 otherwise.
Option last returns the index of the last occurrence
of the substring.
The i_skipToPos argument starts search from the
specified position in the source string,
e.g. Index("words words","word",3) returns 7 .
If i_skipToPos is negative, it specifies the number of characters
from the end of the string in which the search is performed.
Examples:
Another example in which we output all positions of all
-"xxx.." stretches in a sequence " xxxx xxxxx xxxx ... xxxx "
(must end with space)
show Index("asdf","sd") # returns 2
show Index("asdf" "wer") # return 0
a=Sequence("AGCTTAGACCGCGGAATAAGCCTA")
show Index(a "AATAAA") # polyadenylation signal
show Index(a "CT" last) # returns 22
show Index(a "CT" 10) # starts from position 10. returns 22
show Index(a "CT", -10) # search only the last 10 positionsEX = "xxxx xxxxxx xxxxxxxxxxxxxx xxxxxx xxx "
sp=0
while(yes)
x=Index(EX "x" sp)
if(x==0) break
sp=Index(EX " " x)
print x sp-1
endwhile Index ( s_source|S_N_source, { s_pattern| S_N_patterns }, exact | simple | regexp )
exact : search for an exact substring or a matching array of substrings in a source string or array
(e.g. Index({"John","Jon"},"J"),exact)}
simple : case-insensitive search for an substring or a matching array of substrings in an array
(e.g. Index({"John","Jon"},"j"),simple)}
regexp : search for a regular expression(s) in a string or array
(e.g. Index({"John","Jon"},"[jJ]+",regexp))}
Index ( T_tableExpression_orSelection ) → I_matchingRows Index ( T_table_with_graphical_selection_or_rows selection ) → I_matchingRows
- returns an integer array of order numbers (indices) of rows selected by
the table expression.
Example in which we find which value of column B corresponds to a value in column A:
group table t {33 22 11} "A" {"a","b","c"} "B"
Index(t.A==22) # returns 2 for 2nd row
#>I
2
t.B[ Index(t.A== 22 )[1] ] # returns B according to A value
b Index ( T_table i_label label ) → I_matchingRowsset label table Label
Index ( S_data unique ) → I_indexes Index ( I_data unique ) → I_indexesiarray containing indexes of unique elements in the data array,
sorted in ascending order.
test Index( {1 7 5 7 2 1 1 5} unique )=={1 2 3 5}
test Index( {"a" "A" "a" "B" "A"} unique )=={1 2 4} Index ( S_data, s_value [reverse] ) → i_FirstOrLastMatchingElement Index ( I_data, i_value [reverse] ) → i_FirstOrLastMatchingElement Index ( rarray, real [reverse] ) → i_FirstOrLastMatchingElement
- returns integer value indicating the first (or last with reverse option) array element number exactly matching the value string or real, or 0 otherwise. To return an array of matches in an array, use the all option (see below).
Examples:
show Index({"Red Dog","Amstal","Jever"}, "Jever") # returns 3
show Index({"Red Dog","Amstal","Jever"}, "Bitburger") # returns 0
show Index({3 ,2, 8},2 ) # returns 2
show Index( 0.3//0.1//0.2//ND//0.5, ND ) # returns 4
show Index( 0.3//0.1//0.2//ND//0.5, 0.1 ) # returns 2 Index ( S_data, s_value all ) → I_matchPositions Index ( I_data, i_value all ) → I_matchPositions Index ( R_data, r_value all ) → I_matchPositions
- returns iarray listing all positions where the value was encountered.
Index ( I_indexes, i_nofElements inverse ) → I_complementarySetOfIndexesshow Index({"A","B","C","B","B"}, "B") # returns {2,4,5}
show Index({1,2,6,4},3) # returns empty iarray
show Index({1,3} 5 inverse) # returns {2,4,5} Index ( alignment, sequence )integer index of an identical sequence in the alignment of 0.
Index ( alignment selection column ) or Index ( alignment rs ) iarray of column positions selected graphically in the alignment.
See also: macro calcSelSimilarity
Index ( object )
- returns integer value of sequential number of the
current object in the molecular object list, or 0 if no
objects loaded. (Note that here object is used as a keyword.)
Examples:
l_commands = no
read pdb "1crn"
read object s_icmhome+"crn"
printf "The object a_crn. is the %d-nd, while ...\n", Index(object)
set object a_1.
printf "the object a_1crn. is the %d-st.\n", Index(object)
- returns cluster centers (current threshold is taken if not specified)
Index ( tree center [r_threshold] )
- returns indices of table rows which are selected in cluster
Index ( tree selection ) Index ( {iarray|rarray|sarray}, {iarray|rarray|sarray} compare )collection object with four fields:
To get the union, use Unique(Sort(a//b))
a = Random(1,100,50 )
b = Random(1,100,60 )
c = Index( a, b, compare )
show a[ c["A"] ] # elements only in 'a'
show a[ c["AB"] ] # overlap
show b[ c["B"] ] # elements only in 'b'
show b[ c["BA"] ] # overlap
printf "The total number of unique elements is %d\n",Nof(c["A"]//c["B"]//c["AB"]) Index ( {iarray|rarray|sarray}, {iarray|rarray|sarray} compare simple [inverse] )iarray of indices of
matched elements (mismatched with inverse option)
Index( {1 2 3} {1 3 3} compare simple ) # returns {1 3}
Index( {1. 2. 0.} {1. 2. ND} compare simple inverse ) # returns {3} Index( X_single_chem1, X_single_chem2 atom map ) → iarrayiarray of the length equal to the number of atoms in the X_single_chem2. Each element of the result array
contains an atom number in X_single_chem1 which corresponds to the atom number == position_in_the_array in ~X_single_chem2
Index( Chemical("CCO"), Chemical("OCC") atom map ) # returns {3 2 1}
Indexx ( { string | sequence }, s_Pattern )
- returns an integer
value indicating the position of the s_Pattern (see
pattern matching)
in the string, or 0 otherwise. Allowed meta-characters are the following:
Examples:
show Indexx("asdf","s[ed.]") # returns 2
show Indexx("asdfff","ff$") # returns 5 (not 4)
show Indexx("asdf" "w?r") # return 0
Insertion ( rs_Fragment, ali_Alignment [, seq_fromAli ][, i_addFlanks ] [{"all"|"nter"|"cter"|"loop"}] )
- returns the residue selection
which form an insertion from the viewpoint of other sequences in the
ali_Alignment. If argument seq_fromAli is given (it must be the
name of a sequence from the alignment), all the other sequences in the alignment
will be ignored and only the pairwise sub-alignment of rs_Fragment and
seq_fromAli will be considered. The alignment must be
linked
to the object. With this function (see also
Deletion( )
function) one can easily and quickly visualize all indels in the three-dimensional
structure. The default i_addFlanks parameter is 0.
String options:
Examples:
read pdb "1phc.a/" # read the first molecule form this pdb-file
read pdb "2hpd.a/" # do the same for the second molecule
make sequences a_*. # you may also read the sequence and
# the alignment from a file
aaa=Align( ) # on-line seq. alignment.
# You may read the edited alignment
# worm representation
assign sstructure a_*. "_"
display ribbon
link a_*. aaa # establish connection between sequences and 3D obj.
superimpose a_1. a_2. aaa
display ribbon a_*.
color a_1. ribbon green
color ribbon Insertion(a_1.1 aaa) magenta
color ribbon Insertion(a_2.1 aaa) red
show aaa
Info ( [ string ] )
- returns the string with the content previous ICM Info message.
Info ( display )
- returns the string with commands needed to restore the graphics view and the background color.
See also: View () , write object auto or write object display=yes .
Info ( term [map|mmff] ) string with energy terms. E.g.
s_oldterms = Info(term)
..
set terms only s_oldtermsmap is specified, ICM starts looking for m_gc, m_ge, .. etc. maps
and adds a corresponding term. E.g.
s_termsAccordingToExistingMaps = Info(term map)mmff is specified, ICM will select the correct set of the mmff terms.
returns Info ( images ) sarray with advanced details of images, such as their file format (JPEG, PNG, etc.), dimensions,
color space (e. g. RGB, grayscale), transparency, etc.
Info ( predModel ) Info ( s_builtInModelName model ) collection with model properties: type, weights, constant, etc..
Info( "MolLogP" model )
Integer ( l_value )
- returns 0 or 1.
Integer ( r_toBeRounded )
- returns the integer nearest to real r_toBeRounded in the direction
of the prevailing rounding mode magnitude of the real argument.
- converts string into Integer ( string ) integer, ignores irrelevant tail. see also Tointeger
Reports error if conversion is impossible.
Examples:
show Integer(2.2), Integer(-3.1) # 2 and -3
jj=Integer("256aaa") # jj will be equal to 256Iarray( ), Tointeger( )
Integral ( I | R ) iarray (or rarray) of the same dimension containing partial sums (from 1 to i )
of the element in the source array. E.g. Integral({2.,2.,2.}) will return 2.,4.,6.
Integral ( R r_xIncrement ) rarray of the function represented by rarray Ron the periodically incremented abscissa x with the step of r_xIncrement.
Note the difference between this and the above function of partial sums.
The explicit increment form of the function will do the following
E.g. Integral({2.,4.,2.},1.) will return 0.,3.,6.,
while Integral({2.,4.,2.}) will return partial sums 2.,6.,8.
Integral ( R_Y R_X ) rarray of the function represented by R_Y on the set of abscissa
values R_X.
Examples:
Let us integrate 3*x2-1,
determined on the rarray of unevenly spaced x.
The expected integral function is x3-x
# Let us integrate sqrt(x)
x=Rarray( 1000 0. 10. )
plot x Integral( Sqrt(x) 10./1000. ) grid {0.,10.,1.,5.,0.,25.,1.,5.} display
# Let us integrate x*sin(x). Note that Sin expects the argument in degrees
x=Rarray( 1000 0. 4.*Pi )
# 1000 points in the [0.,4*Pi] interval
plot x Integral( x*Sin(x*180./Pi) x[2]-x[1] ) \
{0., 15., 1., 5., -15., 10., 1., 5. } grid display
# x[2]-x[1] is just the incrementx=Rarray(100 ,-.9999, .9999 )
x=x*x*x
plot display x Integral((3*x*x-1.) x) cross
Interrupt
- returns logical yes if ICM-interrupt (Ctrl-Backslash, ^\) has been received by
the program. Useful in scripts and macros.
Examples:
if (Error | Interrupt) returninterruptAction preference, e.g.
interruptAction = "break all loops"
#or
interruptAction = "exit macro"
- returns the Label ( g ) → s string label of the grob. See also: set grob s_label .
-returns Label ( as ) sarray of atom labels which will be displayed. Normally they are atom names. The custom labels can be set with the
set atom label command.
Label ( rs )
- returns sarray of residue labels of the selected residues
rs_ composed according to the resLabelStyle preference , e.g.
{ "Ala 13","Gly 14"}
See also: Name function (returns residue names), and Sarray( rs [append|name|residue]) function
returning selection strings.
Label ( os_objects )
- returns sarray of long names of selected objects.
See also: Name
function which returns the regular object names and the most detailed
chemical names of compounds.
Label ( vs_var )
- returns sarray of labels of selected variables.
Examples:
build string "ala his glu lys arg asp"
resLabelStyle = "Ala 5" # other styles also available
aa = Label(a_/2:5) # extract residue name and/or residue number info
show aa # show the created string array
Label ( T_table ) iarray of table row labels (marks) set from the GUI or by set label command
group table t {1 2 3} "A"
set label t 1 index={1,3}
Label(t) Label ( chem chiral ) sarray of chiral labels for the set of compounds.
set label table Index table label , Nof( X chiral [ 0|1|2.. ] )
Length ( { string | matrix | sequence | alignment | profile } )
- returns integer length of specified objects.
Length ( sarray )
- returns iarray with lengths of strings elements of the sarray.
Length ( X_chem1_single i_at1 i_at2 )integer length in bonds between two atoms in a single molecule
Length ( as1_single as2_single )integer length in bonds between two atoms in a single molecule (-1 if atoms are not connected)
Length ( X_chemarray link )iarray with number of bonds in a shortest path between two marked attachment points.
Length ( seqarray )iarray with lengths of sequence parray elements.
Length ( {iarray | rarray } )
- returns the real vector length (distance from the origin
for a specified vector
Sqrt(Sum(I[i]*I[i])) or Sqrt(Sum(R[i]*R[i])), respectively).
Examples:
len=Length("asdfg") # len is equal to 5
a=Matrix(2,4) # two rows, four columns
nCol=Length(a) # nCol is 4
read profile "prof" # read sequence profile
show Length(prof) # number of residue positions in the profile
vlen=Length({1 1 1}) # returns 1.732051Nof
GaussFit( R_X , R_Y )
- returns a 3-element rarray a,b,c
of the parameters of the Gauss function c*Exp(-(x-a)*(x-a)/b)
read table mol s_icmhome+ "/moledit/Dictionary.sdf" name="Xdict"
add column Xdict function="MolWeight(mol)" index=2 name="molWeight" append format="%.3f"
h = Histogram(Xdict.molWeight,40) # 40 bins
h[?,2] = h[?,2] / Nof( Xdict ) # Normalize by number of molecules
x = h[?,1]
y = h[?,2]
params = GaussFit( x, y )
printf "Best fit: %f * Exp( -(x-%f)*(x-%f)/%f )\n", params[3],params[1],params[1],params[2]
add column tt x y R_out
make plot tt "x=A;y=C;color=#ff0000;size=6.;style=connected;depth=5.;;x=A;y=B;color=#0000ff;size=6.;style=vertical bars;;"
LinearFit( R_X , R_Y , [ R_Errors] )
- returns a 4-element rarray A,B,StdDev,Corr
of the parameters of the linear regression for a scatter plot Y(X):
R_Y = A*R_X + B , where the slope A and the intercept B are the first
and the second elements, respectively. The third element is the
standard deviation of the regression, and the fourth is the correlation coefficient.
Residuals R_Y - ( A*R_X + B) are stored in the R_out array.
You can also provide an array of expected errors of R_Y .
In this case the weighted sum of squared differences will be
optimized. The weights will be calculated as:
Wi = 1/R_Errorsi 2
Example:
X = Random(1., 10., 10)
Y = 2.*X + 3. + Random(-0.1, 0.1, 10)
lfit = LinearFit(X Y)
printf "Y = %.2f*X + %.2f\n", lfit[1],lfit[2]
printf "s.d. = %.2f; r = %.3f\n", lfit[3],lfit[4]
show column X, Y, X*lfit[1]+lfit[2], R_out
A more complex linear fit between a target set of Yi , i=1:n values
and several parameters Xi,j (i=1:n,j=1:m)
potentially correlating with Yi is achieved in 3 steps:
M1=Transpose(X)*X
M2=Power(M1,-1)
W =(M2*Transpose(X))*Y
The result of this operation is vector of weights W for each of m
components.
Now you can subtract the predicted variation from the initial
vector ( Y2 = Y - X*W ) and redo the calculation to find W2 , etc.
A proper way of doing it, however, is to calculate the eigenvalues
of the covariance matrix.
LinearModel( T_weights )sarray called "name" with column names,
and rarray "w" with weights.
It may also have a real header "b" specifying the free term
(the default value is 0.).
model weight function for other regression models may be used as input for LinearModel.
So it is possible to obtain weights from a PLS model,
refine or simplify them, and create a new linear regression model:
n = 1000
add column T Random(-10., 10., n) name="A"
add column T Random(-10., 10., n) name="B"
add column T Random(-10., 10., n) name="C"
add column T T.A + 10.*T.B - 5.*T.C name="Y"
learn T.Y type="plsRegression" name="Y"
Y1 = LinearModel( Table( Y term ) )
predict T Y1# Build model
add column WT {"A", "B", "C"} name="name"
add column WT {0.7, 2.3, -10.} name="w"
add header WT 5.6 name="b"
Y = LinearModel( WT )
# Predict
n = 100
add column T Random(-10., 10., n) name="A"
add column T Random(-10., 10., n) name="B"
add column T Random(-10., 10., n) name="C"
predict T YTable model , predict , learn
- returns the Log ( real ) real
natural logarithm of a specified positive argument.
- returns the
Log ( real r_realBase) real
logarithm of a specified positive argument
(e.g. the base 10 logarithm is Log(x, 10)).
- returns an Log ( rarray ) rarray
of natural logarithms of the array components (they must not be negative,
zeroes are treated as the least positive real number, ca. 10-38).
- returns an Log ( rarray r_realBase ) rarray
of logarithms of the array components (they must not be negative),
arbitrary base.
- returns a
Log ( matrix [ r_realBase ] ) matrix
of logarithms of the matrix components (they must not be negative).
Examples:
print Log(2.) # prints 0.693147
print Log(10000, 10) # decimal logarithm
print Log({1.,3.,9.}, Sqrt(3.)) # {0. 2. 4.}Power
- returns Map( m_map cell ) map in the limits of the crystallographic cell (a,b,c,alpha,beta,gamma).
The source map needs to be equal in size or greater to the asymmetric unit of the cell.
This function helps to prepare local maps for real space refinement
(see make map potential m R_6box )
- returns Map( m_map , I_6box [ simple ] ) map which is a transformation (expansion or reduction) of the input m_map
to new I_6box box ({ iMinX,jMinY,kMinZ,iMaxX,jMaxY,kMaxZ}).
Note that the order of axes in most crystallographic is defined by the MAPS,MAPR,MAPS
parameters and is not always x,y,z. The correctly ordered index is returned by the
Index(
returns a Map( m_map , as ) map around selected atoms . The index box of this selection is returned by the
Index(
Examples:
See also : read object "crn"
read map "crn"
display a_//ca,c,n m_crn
m1 = Map(m_crn, {0 0 0 22 38 38}) # half of the m_crn
m2 = Map(m_crn, {0 0 0 88 38 38}) # double of the m_crn
display m1
display m2make grob map to generate contour around a particular selection
Mass( as | rs | ms | os )
- returns rarray of masses of selected atoms, residues, molecules or objects,
depending on the selection level.
Examples:
build string "ala his trp glu"
objmasses = Mass( a_*. )
molmasses = Mass( a_* )
resmasses = Mass( a_/* )
masses=Mass( a_//!?vt* ) # array of masses of nonvirtual atoms
molweight = Mass( a_1 )[1] # mol.weight of the 1st molecule
molweight = Sum(Mass( a_1//* )) # another way to calculate 1st mol. weight
See also: Nof( sel atom ), Charge( sel ) , Moment ( sel | X_ ) (principal moments of inertia)
Moment( as_nObj|X_n [ pca | simple | all ] ) parray of chemicals (see Chemical ).
Options:
Note: for a linear molecule, the third component of the moment of inertia will be zero.
pca (or no option) : the function returns an array with 3*nObject elements with 3 principal moments of inertia for the selection in each object
simple : the function returns an array with nObject elements with the largest principal moments of inertia (out of three) for the selection in each object
all : the function returns an array with nObject elements with the product of square roots of the principal moments of inertia for the selection in each object. For linear molecules Ixx==Iyy and Izz=0.. In that case the function returns Ixx rather than sqrt(Ixx*Iyy*Izz)build string "ASD"
build string "G"
Moment(a_*.//* ) # three components for each object
3470.9 # first object
2886.5
855.1
167.5 # second object
124.9
48.4
Moment(a_*.//* simple ) # two largest moments of inertia
3470.980225
167.546844
Moment(a_1./2:3/ca pca ) # just two atoms: a linear molecule
86.0
86.0
0.0
Match( s_where s_regexp [i_field=0 [i_startPos=1]] ) → s_matchid_sw = Match(swissEntryHtmlLine, "<DT><A HREF=\"/uniprot/(.+)\">(.+)</A> \(<b>.+</b>\)<DD>(.+)" 1)
namesw = Match(swissEntryHtmlLine, "<DT><A HREF=\"/uniprot/(.+)\">(.+)</A> \(<b>.+</b>\)<DD>(.+)" 2)
descsw = Match(swissEntryHtmlLine, "<DT><A HREF=\"/uniprot/(.+)\">(.+)</A> \(<b>.+</b>\)<DD>(.+)" 3)regexp syntax for the full description of the rules.
Some important hints: add question mark (?) to the end of a matching expression to make the match minimal (to the closest separator).
Without '?' the match will be greedy i.e. it long for the longest match. Example:
Match( "bla = stuff; and more", "=\s+(.*?)\s",1) # ? for minimal
stuff;
Match( "bla = stuff; and more", "=\s+(.*)\s",1) # greedy match
stuff; and"(?i)" or the "(?-i)" prefix
(see also regexp syntax and simple expressions )
Example:
s= "Some text\n Smiles = C1CCCC1 \nmore text"
Match(s,"(?i)smiles\s+=\s*(.+?)\s",1)
# ? in (.+?) means the minimal match, 1 refers to the (..) expression
C1CCCC1 Match( all s_where s_regexp [i_field=0 [i_startPos=1] ) → S_matches Match( S_where s_regexp [i_field=0 [I_startPos={1,..}]] ) → S_matches
- returns Matrix( i_NofRows, i_NofColumns [ r_value] ) matrix of specified dimensions. All components are set to zero or r_value if specified.
Matrix( i_n [ R_n_diagonal ] ) Matrix( i_n [ R_m_row ] ) matrix of specified size. A matching array of diagonal values can be provided.
If the array size does is not equal to i_n , a matrix with i_n rows with R_m_row values will be returned.
Example:
Matrix(3,{1. 2. 3.})
#>M
1. 0. 0.
0. 2. 0.
0. 0. 3.
Matrix(3,{1. 2.})
#>M
1. 2.
1. 2.
1. 2. Matrix( nRows [ R_row ] ) Nof( R_row ) . Example: Matrix(10, {1. 2. 3.})
- converts vector[1:n] to one-row Matrix( rarray [ n ] ) matrix[1:1,1:n].
If you provide a positive integer argument, the input rarray will be
divided into rows of length n. If the argument is negative, it will be split
into columns of length n. Examples,
Matrix({1. 2. 3. 4. 5. 6.},3)
#>M
1. 2. 3.
4. 5. 6.
Matrix({1. 2. 3. 4. 5. 6.},-3)
#>M
1. 4.
2. 5.
3. 6.
Matrix({1. 2. 3. 4. 5. 6.},4)
Error> non-matching dimension [4] and vector size [6]
Matrix( M_square i_rowFrom i_rowTo i_colFrom i_colTo ) → M Matrix( Matrix(3) 0,0,1, 2) # first two columns
- generate a symmetric matrix by duplicating the left or the right triangle of initial square matrix.
Example:
Matrix( M_square { left | right } ) icm/def> m
#>M m
1. 0. 0.
0. 1. 0.
7. 0. 7.
icm/def> Matrix( m right )
#>M
1. 0. 0.
0. 1. 0.
0. 0. 7.
icm/def> Matrix( m left )
#>M
1. 0. 7.
0. 1. 0.
7. 0. 7.
Matrix( S_nHexcolors rgb | color ) rgb (red, green, blue) values. With option color it adds three additional columns for
Example:
This matrix can also be used to calculate a distance matrix and cluster colors, e.g.
makeColorTable # this macro calls the Matrix( .. color ) function
#
Matrix( {"#FFFFAA","#ACBB01"} rgb )
Matrix( {"#FFFFAA","#ACBB01"} color )makeColorTable # create a table
add header icmColors Distance(Matrix(icmColors.Color rgb) ) name="dm"
# click on the cluster tool Matrix( comp_matrix s_newResOrder ) icm/def> Matrix(comp_matrix "CAR")
#>M
2.552272 0.110968 -0.488261
0.110968 0.532648 -0.133162
-0.488261 -0.133162 1.043102 Matrix ( T [ S_colnames ] ) → Madd column t {1 2} {3 4} {4 5} # columsn .A .B .C
M = Matrix( t ) # 3x2 matrix
mm = Matrix( t {"B","C"} ) # 2x2 matrix with .B and .C
Matrix( R_A R_B )
- returns tensor product of two vectors or arbitrary dimensions:
M_ij = R_A[i]*R_B[j]
Examples:
mm=Matrix(2,4) # create empty matrix with 2 rows and 4 columns
mm=Matrix(2,4,-5.) # as above but all elements are set to -5.
show Matrix(3) # a unit matrix [1:3,1:3] with diagonal
# elements equal to 1.
a=Matrix({1. 3. 5. 6.}) # create one row matrix [1:1,1:4 ]
Matrix({1.,0.},{0.,1.}) # tensor product
#>M
0. 1.
0. 0.
Matrix ( rs_1 rs_2 ) matrix of contact areas.
See also: Cad, Area .
Matrix ( ali ) matrix of normalized pairwise Dayhoff evolutionary distances
between the sequences in alignment ali_
(for similar sequences it is equal to the fraction mismatches).
- returns a Matrix ( ali, number ) matrix of alignment. It contains reference residue
numbers for each sequence in the alignment, or -1 for the gaps.
The first residue has the reference number of 0
(make sure to add 1 to access it from the shell).
- returns values generated by the make boundary command for each atom.
Matrix ( boundary )
- returns distance matrix of Matrix ( stack ) stack conformations according to the
compare command and the vicinity parameter. Used for clustering of the stack
conformations.
- retuns 2D histogram of X and Y values.
The R_ruler array consists of limits for X and Y
and step sizes for X and Y and optional bin sizes: {xFrom, xTo, yFrom, yTo, [xStep, yStep] } .
Returned values:
Matrix( R_Xn R_Yn R_ruler )
R_out : contains ruler and actual bin sizes: icm/def> Matrix(Random(0. 5. 20) Random(0. 5. 20) {0. 5. 0. 5. 1. 1.})
#>M
1. 0. 2. 1. 0.
1. 1. 1. 1. 2.
0. 2. 1. 1. 0.
0. 1. 0. 0. 1.
1. 0. 0. 1. 2. Matrix( grob wire ) → M_one_or_large_number
g = Grob("cell",{1. 2. 3.}) # a box with 8 vertices
g = Matrix(g wire)
16777216. 1. 1. 16777216. 1. 16777216. 16777216. 16777216. 1. 16777216. 16777216. 1. 16777216. 1. 16777216. 16777216. 1. 16777216. 16777216. 1. 16777216. 16777216. 1. 16777216. 16777216. 1. 1. 16777216. 16777216. 16777216. 16777216. 1. 1. 16777216. 16777216. 16777216. 16777216. 1. 1. 16777216. 16777216. 1. 16777216. 16777216. 1. 16777216. 16777216. 1. 16777216. 16777216. 1. 16777216. 1. 16777216. 16777216. 1. 16777216. 16777216. 16777216. 1. 16777216. 1. 1. 16777216.
Max ( { rarray | map } )
- returns the real maximum-value element of a specified object
- returns the Max ( iarray ) integer maximum-value element of the iarray.
- returns the Max ( sarray ) string longest common prefix of elements of sarray.
- returns the Max ( R1_n R2_n ) → R_max_nrarray of maximal values.
- returns the Max ( index { iarray | rarray } ) integer index of the maximum-value element of the array (or one of them if many).
- returns maximal distance of the root node
Max ( clusterObject )cl = Split( t.cluster, Max( t.cluster )/2 )
- returns the Max ( index { iarray | rarray } group I_clusterNumbers ) iarray of indices of maximal values, e.g.
see also Max(index { 1. 3. 1. 2. 5.} group { 1 2 1 2 2} )
#>I
1
4 # the maximal element 2. has index 4Min(.. ) and the group .. command.
- returns the Max ( matrix ) rarray of maximum-value element of each column of the matrix.
To find the maximum value use the function twice ( Max(Max( m)) )
- returns the matrix with the larger values of the two input matrices of the same dimensions.
Max ( matrix_nm matrix_nm ) → M_max_nm
- returns the largest Max ( integer1, integer2, ... ) integer argument.
- returns the largest Max ( real1, real2, ... ) real argument.
returns the maximal trailing number in array elements consisting of the s_leadingString and a number.
If there are no numbers, returns Max ( S s_leadingString ) 0.
E.g. Max({"a1","a3","a5"},"a") returns 5.
Max( *grob | *macro | *sequence | *alignment | *profile | *table | *map )
returns the maximal number of shell objects of the specified class. To increase this
shell limit, modify the icm.cfg file.
returns the maximal number appended to grob names:
Max ( grob s_leadingString )
This function is equivalent to Max( Name(grob), s_leadingString ) (see the previous function).
g_skin_1
g_skin_2
Examples:
show Max({2. 4. 7. 4.}) # 7. will be shown
- returns the recommended value of Max( image graphic )GRAPHICS.quality to be used with commands which generate images.
write image memory GRAPHICS.quality=Max(image graphic)
MaxHKL( { map | os | [ R_6CellParameters ] }, r_minResolution ) → I_3hkl_limits
the function extracts the cell parameters from map_ , os_ object, or
reall array of {a,b,c,alpha,beta,gamma}, and calculates an iarray of three maximal
crystallographic indices { hMax , kMax , lMax } corresponding to the specified
r_minResolution .
Median ( { rarray | iarray } )
- returns the real median-value of elements of the specified ICM-shell objects.
print Median(Count(100)) # returns 50.5
Mean ( { rarray | map } )
- returns the real average-value of elements of the specified ICM-shell objects.
Mean ( iarray )
- returns the real average-value of the elements of the iarray.
Mean ( matrix )
- returns rarray [1:m] of average values for each i-th column matrix[1:n,i].
Mean ( R1 R2 )
- for two real arrays of the same size returns rarray [1:m] of average values for each
pair of corresponding elements.
Examples:
print Mean({1,2,3}) # returns 2.
show Mean(Xyz(a_2/2:8)) # shows {x y z} vector of geometric
# center of the selected atoms
Mean({1. 2. 3.} {2. 3. 4.})
#>R
1.5
2.5
3.5
- returns the Min ( { rarray | map } ) real minimum value element of a specified object
- returns the Min ( index { iarray | rarray } ) integer index of the minimum-value element of the array (or one of them if many).
- returns the Min ( index { iarray | rarray } group I_clusterNumbers ) iarray of indices of minimal values, e.g.
see also Min( index { 1. 3. 1. 2. 5.} group { 1 2 1 2 2} )
#>I
1
4 # the minimal element 2. has index 4Max(.. ) and the group .. command.
- returns the Min ( iarray ) integer minimum-value element of the iarray.
- returns the Min ( R1_n R2_n ) → R_min_nrarray of minimum values.
- returns the Min ( matrix ) rarray of minimum-value element of each column of the matrix.
To find the minimum value use the function twice (e.g. Min(Min(m)) )
- returns the matrix with the smaller values of the two input matrices of the same dimensions.
Min ( matrix_nm matrix_nm ) → M_min_nm
- returns the smallest Min ( integer1, integer2 ... ) integer argument.
- returns the smallest Min ( real1, real2, ... ) real argument.
Examples:
show Min({2. 4. 7. 4.}) # 2. will be shown
show Min(2., 4., 7., 4.) # 2. will be shown Min ( alignment, sequence ) → i_nearestSeqName function. Example:
read alignment s_icmhome+"sh3"
b = Sequence("KKYAKAKYDFVARNSSELSMKDDVLELILDD") # like Eps8 seq
iseq = Min(sh3, b) # returns 3.
nam = Name(sh3)[iseq] # "Eps8" is the closest sequence
show $nam
- returns
a Money ( { i_amount | r_amount}, [ s_format] ) string with the traditionally decorated money figure.
s_format contains the figure format and the accompanying symbols.
%m specification for the rounded integral
amount;
%.m specification to add cents after dot. The default is "$%.m", i.e.
Money(1222.33) returns $1,222.33.
%M the same as %m but with dot instead of comma in the European style
%.M the same as %.m dot and comma are inverted in the European style
Examples:
Money(1452.39) # returns "$1,452.39"
Money(1452.39,"DM %m") # returns "DM 1,452"
Money(1452.39,"%.M FF") # inverts comma and dot "1.452,39 FF"
Remainder() function:
function description example
Mod(x,y) brings x to [0 , y] range Mod(17.,10.) → 7.
Remainder(x,y) brings x to [-y/2 , y/2] range Remainder(17.,10.) → -3.
- returns Mod ( i_divisor, [ i_divider ] ) integer remainder.
- returns the
Mod ( r_divisor, [ r_divider ] ) real
remainder r = x - n*y where n is the integer nearest the exact value of x/y;
r belongs to [ 0, |y| ] range.
- returns the
Mod ( iarray, [ i_divider ] ) iarray
of remainders (see the previous definition).
- returns the
Mod ( rarray, [ r_divider ] ) rarray
of remainders (see the previous definition).
The default divider is 360. (or 360) since we mostly deal with angles.
Examples:
phi = Mod(phi) # transform angle to [0., 360.] range
a = Mod(17,10) # returns 7
- selects
Mol ( { os | rs | as } ) molecules
related to the specified objects
os_
, residues
rs_
or
atoms
as_,
respectively.
Mol function to return a mol/sdf formatted string .
The up-to-date version of this function is String( X )
Examples:
See also: show Mol( Sphere(a_1//* 4.) )
# molecules within a 4 A vicinity of the first one
# Sphere function Sphere(as_atoms) selects atoms.Atom, Res, Obj .
- returns empty string.
Name ( )
- returns file name sub- Name ( s|S_Path_and_Name ) → s_name|S_namestring (or array of substrings) if full path is specified , example: Path({"a/b/aa.icb"}) returns {"aa"}
See also Path and Ext
Opions:
Name( s_hint [ simple | unique | object ] )
Examples:
simple : removes non-alpha-numeric symbols from a string and replaces them by underscore.
object : finds a name for a new object starting from s_hint and ensures its uniqueness
unique : checks if the name s_ exists in the ICM-shell. If the name does not exist, it is returned
without changes, otherwise a number is appended to the name to guarantee its uniqueness.
Unique molecular object names and unique molecule names in a given objectName( " %^23 a 2,3 xreno-77-butadien" simple)
23_a_2_3_xreno_77_butadien
a=1
Name("a",unique)
a1
returns a modified input string if an object with suggested name already exists, or the input string if it is unique already. E.g. Name( "target", object, unique ) will return "target" or "target_1" if a_target. exists already.
Name( s_obj_name_hint object unique )
returns a unique name for a new molecule in existing object os1_object , e.g.
Name( "a", a_2. unique) will return "a" or "a1" if Name( s_mol_name_hint os1_object unique ) a_2.a already exists
Name( variable any_shell_variable )string with a name of a provided shell variable.
add column t {1 2 3} name="A"
Name( variable t )
Name( variable t.A ) Name ( className ) → S_names command,function,macro,integer,real,string,logical,iarray,rarray,sarray,matrix,map,grob,alignment,table,profile,sequence
returns the list of html documents or scripts in ICM shell
Name( string [ html | command ] )
- returns a string array of names of selected objects for the specified class.
Name ( { iarray | rarray | sarray | matrix | map | grob | alignment | table | profile | sequence | chemical | reaction | slide | model | tree } selection )
- returns Name ( as [full] ) sarray of names of selected atoms,residues,molecules or objects.
With option full it returns sarray of the selection expressions (e.g. {'1abc.a/13/ca'} ), one item per array element. Name( .. full ) function is often used to form columns of clickable cells.
Example:
read pdb "1zzz"//"3zzz"
add column t Name(a_*.H full) name="A"
set format t.A "<!--icmscript name=\"1\"\ndsSelection \"%1\" --><a href=#_>%1</a>"String( as ); Sarray( as )
- returns Name ( as|rs|ms|os field ) sarray of unique names of assigned tags (fields), see also set field name .
- returns Name ( as sequence ) sarray of chemical names of the selected atoms according to the icm.cod
file (one-letter chemical atom names are low case, e.g. "c", two-letter
names start from an upper-case letter, like "Ca"). The names from the
periodic table are used in the wrGaussian macro.
- returns Name ( rs ) sarray of names of selected residues.
To obtain a one-letter code sequence, use Sequence( rs_ )
and to convert it to a string use String( Sequence( rs_ )) .
- returns Name ( ms ) sarray of names of selected molecules.
- returns Name ( ms chain ) sarray of chain names of selected molecules.
- returns Name ( chem_array ) sarray of names of chemicals in an array ( see also )
- returns Name ( ms sequence ) sarray of names of sequence linked to the specified molecules ms_ or empty strings.
- returns Name ( ms alignment ) sarray of names of alignments linked to the specified molecules ms_ or empty strings
- returns Name ( ms swiss ) sarray of names of swissprot names corresponding to the specified molecules ms_ or empty strings . See also the set swiss command.
- returns Name( os ) sarray of names of selected objects. E.g. Name( a_ )[1] returns string with the name of the current object.
- returns Name ( vs ) sarray of names of selected variables.
- returns Name ( alignment ) sarray of constituent sequence names.
- returns Name ( table ) sarray or constituent table ICM-shell object names.
- returns Name ( sequence ) string name of specified sequence.
- returns Name( T column )sarray of column names
- returns Name( T header )sarray of header names
- returns Name( T selection )sarray of selected (in GUI) column names
- returns Name( collection )sarray of keys of the collection
- returns Name( collection s_filter )sarray of keys of the collection which satisfy the s_filter expression. s_filter can be any logical
expression which operates with key or value or both.
c = Collection( "a" yes, "b" no, "c" yes )
Name( c "value==yes" ) # returns only "a" and "c"
Name( c "value" ) # the same as above
Name( c "!value" ) # "b" Name( gui {html|table|alignment} )sarray of shell objects in the order of their tabs appear in the GUI. Notice that the order of
tabs corresponding to html-documents, tables or alignments can be changed with drag and drop.
It will lead to a different order retuned by the Name function.
-returns name of the currently active object (the active tab) in the class.
Name( foreground {html|table|alignment|slide} )read alignment msf s_icmhome+"azurins" # load alignment
seqnames = Name(azurins) # extract sequence names
show Name( Acc( a_/* ) ) # array of names of exposed residues
Name( chemical property )sarray of names of loaded descriptors/models (e.g. "MolLogP") for chemicals.
Name( soapStruct ) SOAP services for further information.
Name( ali seq ) → s_nameOfTheClosestSequenceread alignment s_icmhome+"sh3" # alignment
readUniprotWeb "FYN_HUMAN"
Name(sh3,FYN_HUMAN) # returns "Fyn"
- returns Name( string html ) sarray with the names of all the HTML objects in the project
- returns Name( string command ) sarray with the names of all the scripts in the project
- returns Name( tree-parray i_parrayIndex [index|label|matrix|sort|split] )string names of different properties of the tree cluster object.
The following names can be returned:
Name( T.cluster 1 ) returns the tree name shown in GUI
index : returns the name of a table column containing the unique index number of each row
in the order of the data tree (compare with the split option which returns the branch number).
a = "T."+Name(T.cluster 1 index)
sort $a # sorts rows in the tree orderlabel : returns the format string of the tree node label, e.g. "%NAME(%ID)", referring to table columns
called t.Name and t.ID. The names need to end with a separator or semicolumn.
matrix : if the distance matrix was used when making a tree,
and this matrix was attached to the table header, the shell name of this distance matrix is returned.
Example:
read table "t.tab"
make tree t matrix "upgma" # attaches distance matrix to the table header
show Name(t.cluster 1 matrix)sort : returns the column name which was used to additionally order the data tree during the tree construction.
split : returns the column name in which the branch order number (at a certain fixed split level) is stored.
Name( chemarray )sarray of names of chemarray.set name command.
set name other chemical functions
Name( conf ) → comments for the global stack Name( os conf ) → comments for the embedded stack of the object Name( seq_parray ) → S_namessarray with stored names of sequence parray elements
set name sequence
Name( predModel column )sarray with column names or chemical fingerprint chain information.
Descriptor function.
Name( myModel column )
#>S string_array
[#6;H3]
[#6;H2]-[#6;H3]
[#6;H2]
...Descriptor
Name( object_parray )sarray with names of objects in the object_parrayobject parray
Name( imageArray )sarray with names of images
image parray
Name( sql )string, which can be used as the connect= option with almost all Molcart commands
to specify the connection. See also molcart connection options
Name( sql database )sarray listing all databases in the current Molcart connection
Name( sql table [s_database] )sarray listing all tables in the current or specified database
Name( molcart table [s_database] )sarray listing chemical tables in the current or specified database
Name( sql connect )sarray with the connection parameters stored in user's settings: {host,user,password,database}
Name( s_dbtable sql column )sarray listing column names in the specified table. Table name may be prefix with database name with dot.
molcart, Type molcart, Nof molcart
Namex ( os|ms|rs ) → S_comments
Namex ( s_MultiObjectFile )
- returns sarray of comments of selected objects os_
(i.e. a string for each object). This field is set to the
chemical compound name by the
read pdb command. Alternatively, you can
set your own comment with the
set comment os_ s_comment
command.
If you have a single object and want to convert
a string array of one element (corresponding to this one object) to a
simple string, use this expression, e.g.: Sum(Namex(a_)).
Other manipulations with a multiline string can be performed with the
Field,
Integer,
Real,
Split functions (see also
s_fieldDelimiter).
Example. We stored values in the comment field in annotations like this: "LogP 4.3\n". Now we extract the values following the "LogP" field name:
remarks = Namex( s_icmhome+"log3.ob") # get remarks directly without reading
group table t Rarray(Field(remarks,"vacuum",1,"\n")) "vacuum"
group table t append Rarray(Field(remarks,"hexadecane",1,"\n")) "hex"
show t
read object s_icmhome + "log3" # read multiple object file
# extract numbers following the 'LogP' word in the object comments
logPs = Rarray(Namex(a_*.),"LogP",1," \n")set comment
- returns Namex ( seq ) string of long name ('description' field in Swissprot).
- returns Namex ( seqarray ) sarray with long names of sequence parray elements
Example:
read index s_inxDir+"/SWISS.inx"
read sequence SWISS[2] # read the 2nd sequence from Swissprot
show Namex( sequence[0] )
- returns Namex ( imageArray ) sarray with comments associated with images
image parray, Name image .
Next ( { as | rs | ms | os } )
- selects atom, residue, molecule, or object
immediately following the selected one. Next( the_last_element ) returns
an empty selection.
Examples:
read object s_icmhome+"crn.ob"
Next( a_/4 ) # show residues number 5
- selects atoms forming covalent bonds with the selected single atom.
Option Next ( as { bond | tree } ) tree allows one to select only atoms above a given atom in an icm-tree.
hetatm molecule is covalently attached to a polymer:
Nof will return a number larger than 0
read pdb "2vsd"
ms1 = a_a2
l_cov_attached = Nof( Mol(Next( ms1 bond)) & !(ms1) ) > 0
Another example:
build string "his"
display
display a_/his/he2 ball red
display Next( a_/his/he2 bond ) ball magenta # show atom preceding he2
cd2_neigh = Next( a_//cd2 bond )
for i=1,Nof(cd2_neigh)
nei = cd2_neigh[i]
print " Distance between a_//cd2 and ",Sum(Name(nei)), " = ", Distance( a_//cd2 nei)
endfor
Nof( X .. )
Summary:
Nof( P_distArray, distance ) → i_nofDistances # see make distance Nof( X, s|S_smarts ) → I|M_nOfChemMatches Nof( X, 'maxRing'|'minRing'|'maxFusedRings'|'maxFusedAromRings'|'rotBond'|'HBA'|'HBD'|'APO' ) → I_nOfChemMatches Nof( X, atom|chiral|molecule|selection|superimpose|topology ) → n Nof( X, chiral, {0|1|2|3} ) → nChiralCenters of particular type Nof( X, library ) → enumeration_library_size Nof( site rs_|seq_ ) → i_nSites Nof( model vector ) → i_nLatentVectors Nof( drestraint|residue|vrestraint type ) → i Nof( as {bond [error] | selftether [error] } ) → i Nof( rs|ms {atom | tautomer } ) → I #in each res. or mol. Nof( stack ) → the number of stack atoms Nof( os_1, "bio" ) → nBiomol # see: Select|Transform(os "bio" i) Nof( [os_1] stack ) → the number of stack confs Nof( fork ) → the number of CPU cores available Nof( map grid ) → the number of 4D layers in map Nof( g {1|2|3} ) → nVertex|nLines|nTriangles Nof( s s_substring [pattern] ) → nMatches Nof( s_file.ob|.cnf ) → number of objects in .ob, or conformations in .cnf files Nof( tree [i_at=1|all] ) → nEntries Nof( tree [i_at=1] tree|auto ) → nClusters|nSuggestedClusters Nof( idb s_indexedIDBfile ) → nRecords Nof( S|I s|i ) → i_nMatched_el Nof( S s regexp ) → I_nMatches_in_each Nof( s_table_name sql [s_connectionID] ) → nRows Nof( s_table_name molcart unique ) → nUniqueChemicals
Nof ( className )integer number of objects in a class (e.g. Nof(sequence) ). Classes:
iarray,rarray,sarray,sequence,aselection,vselection,alignment,matrix,map,grob,string,object
- returns Nof ( { iarray | rarray | sarray | chemarray | parray } ) integer number of elements in an array.
Note that distanceParrays or hbondParrays returned by the make distance of make hbond commands have a two-level
structure in which the actual list of bonds or distances is the nested to the main level of this parray. Therefore to get
the number of distances or hbonds one needs to use the following function.
- returns the total number of nested atom pairs.
Nof ( hbondChunkArray|distChunkArray distance )
- returns Nof ( ali ) integer number of sequences in a specified alignment ali_
(see also Length( alignment ) ).
- returns Nof ( matrix ) integer number of rows in a matrix (see also Length( matrix) function which returns number of columns).
- returns Nof ( table ) integer number of number of rows in a table
- returns Nof ( map ) integer number of grid points in a map.
- returns Nof ( grob ) integer number of points in graphics object.
- returns Nof( { os | ms | rs | as | vs } ) integer number of selected objects, molecules, residues, atoms or variables respectively.
- returns Nof( { as on|off | bond [error] | selftether [error] ) integer number of atoms that are hidden ( off ) or present ( on ). See also set as on | off . Also counts bonds, bonds and selftethers
- returns Nof( { rs|ms atom | tautomer ) iarray with number of atoms or tautomers in each residue or molecule. Depends on selection levels.
- returns the total Nof ( { atoms | residues | molecules | objects | conf | stack | tether | vrestraint } ) integer number things.
atoms : same as Nof(a_*.//*) , except that Nof(atoms) will work even if the object does not exist.
residues : same as Nof(a_*.*/*)molecules : same as Nof(a_*.*)objects : same as Nof(a_*.)conf : number of confomations (`conf) in the global stack
stack : number of atoms in an object which was used to create a stack , e.g. Nof( a_2. stack )
tether : the total number of tethers in the current object, same as a_//T
vrestraint
- returns the number of available processor cores for spawning new processes. See: Nof ( fork )fork and wait
- returns 1 if the force field parameter library is loaded and 0 otherwise.
Nof ( library )
- returns 1 if the mmff library is loaded and 0 otherwise.
Nof ( library )
- returns the number of active graphical planes
Nof ( plane )
- returns Nof ( site [ ms | seq ] ) integer number of sites in the selected molecule or the current object or sequences.
- returns Nof ( os1 stack ) integer number of conformations in a built-in stack of a specified object.
- returns Nof ( s_objFileName.ob ) or Nof( s_stackFileName.cnf ) integer number of conformations in an object file, or conf in a stack conformation file.
Example:
nObj = Nof("target.ob")
if(nObj >=2) read object "target.ob" number=2
for i=1,Nof("def.cnf") # stack is NOT loaded
read conf i "def.cnf"
endfor
- returns Nof ( os_singleObj stack ) integer number of conformations in the object stack. Note that stack stored in object is not the same as the global shared stack.
E.g.
build string "HWEH"
montecarlo store # creates stack and stores it in object
Nof(a_ stack) # returns the number of conformations in object stack
- returns Nof ( string, substring ) integer number of occurrences of substring in a string.
E.g. Nof("ababab","ba") returns 2
- returns Nof ( string, substring, pattern ) integer number of occurrences of regular pattern in a string.
E.g. Nof("ababab","b?",pattern) returns 2
Example with a strange DNA sequence dn1:
if(Nof(String(dn1),"[!ACGT]" pattern) > 0.5*Length(dn1)) print " Warning> Bad DNA sequence"
Nof ( className selection ) integer number of selected ICM-shell variables.
This selection does not work the following types:
aselection, vselection, string , object .
Examples:
nseq = Nof(sequences) # number of sequences currently loaded
if(Nof(object)==0) return error "No objects loaded"
if ( Nof( sequence selection ) == 2 ) a = Align( selection ) Nof ( {table|alignment|grob} display ) integer number of displayed ICM-shell variables.
Nof( grob display ) # number of meshes displayed in 3D
Nof( table display ) # number of spreadsheets visible in GUI
- returns Nof ( tree [i_index=1] ) integer number of entries in the cluster tree.
- returns Nof ( tree [i_index=1] tree ) integer number of clusters at current split level
- returns an Nof ( tree [i_index=1] auto ) integer guess for a recommended number of clusters
- returns an Nof( chemarray, "ring" ) iarray containing the number of rings in each array element.
show Nof(Chemical("C1CC2CC1CCC2") "ring" )
- returns an Nof( chemarray, "minRing" ) iarray of max ring sizes
- returns an Nof( chemarray, "maxRing" ) iarray of min ring sizes
- returns the number of chiral or racemic centers as follows:
Nof( chemarray, chiral [ 1|2|3 ] )
Example: add column t Nof(t.mol chiral 3) "nRacemicCenters"
Label( X chiral )
Nof( chemarray, s_smarts [group] )iarray of number of matches with the SMART pattern for each element of the chemical array.
group option toggles the special search mode when all atoms in the pattern except the attachment
points needs to match the chemical exactly.
Nof( Chemical("CP(C)(C)=C"), "P~*" )
Nof( Chemical("OP(O)(O)=C"), "P~O" )
Nof( Chemical( "CC(=O)OC1=CCCC=C1C(O)=O" ), "[*;!D1]!@-[*;!D1]" )
# the group option examples
Nof( Chemical("C(=CC=CC1C(=CC=C(C2)C(=CC=CC3)C=3)C=2)C=1") "C(=C1)C=[C*]C=C1" group )[1] # returns 2
Nof( Chemical("C(=CC=CC1C(=CC=C(C2)C(=CC=CC3)C=3)C=2)C=1") "C1C=[C*]C=C[C*]=1" group )[1] # returns 1 other chemical functions SMILES and SMARTS
Nof( distobject distance )distance parrays created by the following commands.
The full information can be exported into a table with the Table(.. distance) function.
make distance to create distance labels;
make hbond for hydrogen bonds;
make angle for planar angles;
make torsion for dihedral angles.
read pdb "1crn"
convertObject a_ yes yes no no
make hbond name="hbonds_crn"
show Nof( hbonds_crn ) # counts distances
- returns the total library size
Nof( X_scaffold library )
- returns Nof( X_scaffold group ) iarray of substituent R-group array sizes
link group , chemical , Chemical .
- counts the number of entries (rows) in an SQL table
Nof( s_dbtable sql [s_connectionID] )
- counts unique chemicals in an SQL table
Nof( s_dbtable molcart unique ) molcart
- returns number of latent vectors of the PLS model
Nof( PLS_model i_num )
- counts numbers of element in complex SOAP object (array or structure)
Nof( soapObject ) SOAP services for information.
- returns the specified norm of the vector.
Supported norm types ( s_type ) are (this parameter is case-insensitive):
Norm ( R_vector s_type )
"range": (default), a difference between the maximal and the minimal value in the vector Max (v) - Min (v)
"rmsd": the root-mean-square-deviation of vector values from their mean value, also returned by the Rmsd (v) function.
"euclidean", a.k.a. "L2": the euclidian distance between the origin and a point defined by the input vector. Also returned by: Sqrt(Sum(Power(v,2))), Length (v)
"cityblock", a.k.a. "L1": the sum of the absolute values of all vector components, Sum(Abs(v))
"Linfinity", "Linf": the largest value in the vector, also returned by Max(Abs( v ))
Norm ( {3., 4.}, "euclidean" )
Norm ( {1. 3. 3. 6. 3. 4. 5.7 7.}, "RMSD" ) # case insensitiveNormalize
Normalize( R, r_f0 r_t0) → R_norm #[f0,t0]→[0.,1.]
change the input range of values to the destination range. If the second array has only two elements the source range is derived from the input array R .
Normalize( R, R2_f_t|R4_f0_t0_f_t) → R_norm #[f0,t0]→[f,t]
- returns the normalized Normalize ( R_vector s_type )rarray (i.e. the input vector) by dividing each vector element by its norm ( s_type can be "range","rmsd","euclidean","citiblock","linf", see more in Norm ). Note that this form of normalization is NOT suitable for
elements of a column in a table.
To standardize data in a table column and remove or soft-trim outliers use Trim
( e.g. linear transformation to a range [0.,1.] is given by Trim( R rainbow ) )
- returns the Normalize ( matrix ) matrix linearly transformed into the [0.,1.] range.
Norm( Normalize( v, s_type ) s_type ) # always returns 1.2.*Normalize ( {3., 4.}, "euclidean" ) #make euclidean length 2.
InList ( S_list S_testedItems ) sarray of S_testedItems elements which are included in the first list.
NotInList ( S_list S_testedItems ) sarray of S_testedItems elements which are not in the first list.
Examples:
NotInList({"cc","aa","bb"} {"aa","dd","ee"}) # returns {"dd","ee"}
InList ({"cc","aa","bb"} {"aa","dd","ee"}) # returns {"aa"}InList( S_list , S_X )
- selects Obj ( { ms | rs | as } ) object(s) related to the specified molecules, residues or atoms, respectively.
Examples:
See also: show Obj( a_*./dod ) # show objects containing heavy waterAtom, Res, Mol .
- returns rarray of occupancy for the specified selection.
If residue selection is given, average residue occupancies are returned.
Occupancy ( { as | rs } )
See also:
set occupancy.
Examples:
read object s_icmhome+"crn.ob"
avO=Min(Occupancy(a_//ca)) # minimal occupancy of Ca-atoms
show Occupancy(a_//!h*) # array of occupancy of heavy atoms
color a_//* Occupancy(a_//*) # color previously displayed atoms
# according to their occupancy
color ribbon a_/A Occupancy(a_/A) # color residues by mean occupancy
- returns the working directory (same as Path ( ) Path(directory).
- returns header sub- Path ( s|S_FullFileName ) string (or sarray) with the path(s).
Example: Path("a/b/c/dd.icb") returns "a/b/c/"
See also Name and Ext
- returns the full file name including absolute directory, and the filename with extension
Path ( s_FullFileName full )
Examples:
See also: sPath=Path("/usr/mitnick/hacker.loot") # returns "/usr/mitnick/"
Path("~/.cshrc" full )
/home/crepe/.cshrcName() and Extension() functions which return two
other components of the full file name.
Path ( indexTable ) → s_sourceFilestring path to the source data file for the indexTable .
The full name is returned by the File function.
Example:
See also: write index mol "/data/chem/nci.sdf" "./nci.inx"
read index "./nci.inx"
Path(nci) # returns location of the source nci.sdf file
/data/chem/write index , File( T_indexTable database )
Path ( origin [ S|s_script_with_args [S_args] ] )
- returns a path to the ICM executable and optional arguments. It is useful when you want to call a named icm-script with arguments in a multi-platform compatible way.
ICM binary can also be found in the Version(full) string.
Path(origin "myicmscript.icm file.icb -v -max=2.3")
/pro/icm/icm/icm myicmscript.icm file.icb -v -max=2.3
Path(origin, "myicmscript.icm",{"file.icb", "-v", "-max=2.3"})
/pro/icm/icm/icm myicmscript.icm file.icb -v -max=2.3
Path(origin, {"myicmscript.icm","file.icb", "-v", "-max=2.3"})
/pro/icm/icm/icm myicmscript.icm file.icb -v -max=2.3
Path ( directory )
- returns the current working directory.
Path ( last )
- returns the path of the last icm-shell script called by ICM.
Path ( preference )
function obsolete
Path ( s_somePath fix )Path("/home/"+"/"+"theuser//" fix ) # == "/home/theuser/"
or
Parray( grob|iarray|object|rarray|sarray|sequence i_n ) Parray( i_n iarray|object|rarray|sarray|sequence) Image function, to store logicals see below.
Extracting the parray elements back to shell objects can be done with one of three methods:
i|r|sarrays , sequences
Image), maps,
Parray ( s_smiles smiles ) Parray ( s_molFileText mol )
add a column of row-arrays. E.g.
Parray ( matrix ) add column t Parray(Matrix(10))
set property plot t.A
- returns an empty Parray ( model s_modelName ) parray of type model It has two reserved fields "type" (set to "Custom" ) and "dim" .
This object also behaves as a collection which can hold additional named elements.
- returns an Parray ( object ) object parray containg all ICM molecular objects loaded
- returns an Parray ( object os [stack] ) object parray of ICM molecular objects from the object selection.
If stack keyword is specified, the current stack is stored into the object.
- returns a Parray ( sequence rs )sequence parray of size 1 containing the residues specified.
- returns a Parray ( sequence [selection] )sequence parray containing all sequences loaded into ICM (with the selection option only the GUI-selected ones).
- returns a Parray ( sequence|object i_n )sequence parray or object parray containing i_n empty objects
read table mol "ex_mol.mol" name="t"
s = String(t.mol[1]) # sss contains mol/sdf text
t.mol[1] = Parray(s mol) # sss is parced and converted
- returns sequence pattern string which can be searched in a single sequence with the
find pattern command or in a database with the
find database pattern=s_pattern command.
If ICM-consensus string s_consensus is provided as an argument, the string
is translated into a regular Pattern ( { s_consensus | ali } [ exact ] ) pattern expression
(e.g. an expression "R+. ..^D" will be translated to "R[KR]?\{3,6\}[ACGS]D" ).
If alignment ali_ is given as an argument, the pattern is either extracted directly from the alignment,
option exact, or is converted to consensus first, and only after that translated into a pattern.
For example, an alignment position with amino acids A and V will be transformed into pattern [AV]
with option exact
and into pattern [AFILMPVW] without the option. Additionally, the exact option
will retain information about the length of the flanking regions.
Example:
read sequence s_icmhome + "zincFing"
group sequence aaa
align aaa
show Pattern("#~???A% ?P") # symbols from consensus string
show Pattern(aaa)
show Pattern(aaa exact)
- returns Pattern ( s_seqPattern prosite | residue ) string containing the prosite -formatted sequence pattern.
The input string s_seqPattern is an ICM sequence pattern .
- returns Pattern ( rs ) → s_res_barcode string "barcode" with selected residues followed by the length of the intervening gaps.
This function can be applied with the 'B' and 'Q' residue selections. E.g.
read pdb "1xbb:
Pattern(Res( Sphere( a_H a_A -1.1)))
A47ME1AE1G46L
a_*.*/BA47ME1AE1G46L
- e.g. Pattern(1crn_a) returns "C??C???...C"-style pattern
Pattern ( seq disulfide ) → s_Cys_pattern
- returns the Pi real value of Pi ( 3.14...).
Examples:
print Pi/2.
- returns Potential ( as_targets as_charges ) rarray of Nof( as_targets) real values of electrostatic potentials
at as_targets atom centers.
Electrostatic potential is calculated from the specified charges
as_charges and the precalculated boundary
(see also REBEL, make boundary
and How to evaluate the pK shift).
Examples:
read object s_icmhome+"crn"
# prepare electrostatic boundary descriptions
make boundary
# potential from oe*, od* at cz of two args
show Potential(a_/arg/cz a_/glu,asp/o?* )
print 0.5*Charge(a_//*)*Potential(a_//* a_//* )
# the total electrostatic energy which is
# actually calculated directly by show energy "el"
- returns Power ( r_base, { r_exp | i_exp} ) real r_baser_exp or r_basei_exp.
Note that r_base may be negative if the exponent is an integer,
otherwise error will be produced.
- returns Power ( r_base, R_exp ) rarray of the r_base taken to the R_exp powers.
- returns Power ( R_base, r_exp ) rarray with each of the R_base elements taken to the r_exp power.
- returns Power ( R_base, R_exp )rarray with R_exp powers of the according R_base elements. Input arrays should have the same size.
- returns Power ( r_base, M_exp ) matrix of the r_base taken to the M_exp powers.
Example:
Power(2.,{1. 2. 3.}) # returns {2.,4.,8.}
- returns Power ( rarray, r_Exponent ) rarray of elements taken to the specified power.
- for square matrix returns the source matrix taken to the specified power.
If the exponent is negative, the function returns the n-th power of the inverse
matrix.
Power ( matrix, integer )
Examples:
size=Power(tot_volume,1./3.) # cubic root
read matrix "LinearEquationsMatrix" # read matrix [1:n,1:n]
read rarray b # read the right-hand column [1:n]
x=Power(LinEquationsMat,-1) * b # solve system of linear equations
a=Rot({0. 1. 0.}, 90.0)
# create rotation matrix around Y axis by 90 degrees
if (Power(a,-1) != Transpose(a)) print "Wrong rotation matrix"
# the inverse should be
# equal to the transposed
rotate a_1 Power(a,3) # a-matrix to the third is
# three consecutive rotationsLog
Predict( chem_array )table with specified chemical properties:
For a specific function, see the following function:
"DrugLikeness : the decision value is returned for each molecule:
negative values for NON-drug-like molecules, positive for the drug like molecules.
"MolArea" : surface area in square Angstroms
"MoldHf" : delta Heat of Formation in [cal/mol]
"MolHalfLife" : estimated half-life in hours (?)
"MolLogP" : the octanol water transfer ( Log(Coct/Cwater) : values over 5. are hydrophobic, poorly soluble molecules, the negative values are polar molecules with likely permeation problems. Optimal range from 1. to 3.
"MolLogS" : solubility in Log( C [in moles/liter ] ). Measures the log of the concentration at which you expect
one half of your molecules in an aggregated form. E.g. -10. means virtually unsoluble, -1. means well soluble.
"MolPSA" : estimated topological Polar Surface Area, predicted absolute polar surface area in square Angstroms.
"Volume" : predicted absolute volume of in cubic Angstroms.
Predict( chem_array {s_property|P_model} )rarray with one of the following properties :
"DrugLikeness","MolArea","MoldHf","MolHalfLife","MolLogP","MolLogS","MolSynth","MolPSA","Volume"
"MolSynth" : common nature of chemical connections. Value of 1. means common, 0. means uncommon.
Name ( chemical property ) function.
Predict(Parray( "C1CCNCC1" ) )
#>T
#>-DrugLikeness-MoldHf------MolLogP-----MolLogS-----MolPSA------Volume-----
-1.006698 -11.227206 0.746186 -0.528251 12.391162 102.286931
read table mol "drugs"
add column drugs Predict(drugs.mol "MolLogP" ) name="MolLogP"
746186
- returns the Probability ( s_seqPattern ) real
probability of the specified sequence pattern. To get mathematical expectation
to find the pattern in a protein of length L, multiply the probability by
L-Length( s_seqPattern).
Examples:
# chance to find residues RGD at a given position
show Probability("RGD")
# a more tricky pattern
show Probability("[!P]?[AG]")
- returns the Probability ( i_minLen, r_Score [, { identity | similarity | comp_matrix | sort } ] ) real expected probability that a given or higher score ( r_Score) might occur
between structurally unrelated proteins (i.e. it is essentially the
probability of an error). This probability can be used to rank the
results of database searches aimed at fold recognition. A better score corresponds
to a lower probability for a given alignment. The four types of scores
are given in the description of the
Score
function. Each score has a different distribution which was
carefully derived from all-to-all comparisons of sequence of protein domains.
identity: the number of identical residues * 100 and divided by the minimal sequence length
similarity: alignment score without gap penalties *100. and divided by the minimal sequence length
comp_matrix: alignment score without gap penalties (unnormalized)
sort: an additional score for ranking
Example:
Probability( 150, 30, identity)*55000. is the mathematical expectation of
the number of structurally unrelated protein chains of 150 residues with
30% or higher sequence identity which can be found in a search through
55000 sequences. The inverse function is
Score .
Probability ( ali_2seq, [ i_windowSize1 i_windowSize2 ] [ local ] ) 
- returns the rarray
of expected probabilities of local insignificance
of the pairwise sequence alignment ali_2seq.
The Karlin and Altschul score probability values (option local ) or
local ZEGA probabilities (see also
Probability(i_, r_))
are calculated
in multiple windows ranging in size from
i_windowSize1 to w_windowSize2 (default
values 5 and 20 residues, respectively). The exact formulae for the Karlin and Altschul
probabilities (option local ) are given in the next section and the ZEGA probabilities
are given in the
Abagyan&Batalov paper.
The window with the lowest probability
value is chosen in each position.
The array returned by this function can be used to color-code the regions of insignificant
sequence-structure alignment in modeling by homology. One can use the
Rarray(R_,ali_,seq_)
function to project the array onto selected sequence.
To calculate an array of mean scores for each column of a multiple sequence alignments use the Rarray( ali [ exact ] ) function.
Example:
read alignment s_icmhome+ "sx" # 2 seq. alignment
read pdb s_icmhome+"x"
p= -Log(Probability(sx))
display ribbon a_
color ribbon a_/A -Rarray(p,sx,cd59)
# Rarray projects the alignment array to the sequence
- returns a dot Probability ( seq_1 seq_2 [ i_windowSize1 i_windowSize2 ] ) matrix of probabilities of local statistical comparison between the two sequences.
This matrix contains local probability values
that two continuous sequence fragments of length ranging from
i_windowSize1 to w_windowSize2 have statistically
insignificant alignment score , which means that the match is random.
Visualization of this matrix allows one to see periodic
patterns if sequence is compared with itself as well as identity
alternative alignments.
The formula is taken from the Karlin and Altschul statistics:
P= 1-exp(-exp(-Lambda*Sum(score in window)/K)), where Lambda and K
are coefficients depending on the
residue comparison matrix.
This example allows one to trace the correct alignment despite an about
100 residue insertion:
read pdb sequence "2mhb"
read pdb sequence "4mbn"
m=Probability(2mhb_a 4mbn_a 7 30)
print " pProbability: Min=" Min(Min(m)) "Max=" Max(Max(m))
PLOT.rainbowStyle="white/rainbow/red"
# show probability of the chance matching (comparable to the BLAST P-value)
plot area m display color={.2 0.001} transparent={0.2 1.} link grid
# OR show -Log10(Probability)
plot area -Log(m,10) display color={0.7 3.} transparent={0. 0.7} link grid
Profile ( chem i_at1 i_at2 i_at3 i_at4 ) Profile ( chem selection ) Profile ( v_tor {group|randomize} )
- creates Profile ( alignment ) profile from an alignment
- returns various advanced Property ( grob option ) grob properties as logical.
full -- dual in-out lighting of the grob
surface -- whether only the front face of the triangles is shown (back faces culled)
texture -- allow textures
material -- allow materials
grid -- (requires texture). Use only the fractional part of the texture coordinates (allows repetitive pattern textures)
heavy -- grob lids on sliced surfaces
See also set property
Putarg( s_name s_value )Getarg .
Putenv ( " s_environmentName = s_environmentValue " )logical yes if the named shell-environment variable is created or modified.
Putenv ( ) Getarg )
into the unix shell as shell arguments. Returns the number of set variables.
To eliminate an agrument from the list, use the
Getenv ( s_argName delete ) function.
Examples:
show Putenv("aaa=bbb") # change/add variable 'aaa' with value 'bbb' to environment
show Getenv("aaa") # check if it has been successfulExistenv, Getenv, Getarg.
- returns the real array of van der Waals radii of atoms in the selection.
Radius ( as )
These radii are used in the construction of the molecular surface
(skin) and can be found (and possibly redefined) in the
icm.vwt file.
- returns the Radius ( as ball ) rarray of the graphical radii of the xstick or ball representation. Normally they are defined by the
GRAPHICS.stickRadius parameter and GRAPHICS.ballStickRatio . They can be set to custom values with the
set atom
- returns the Radius ( as surface ) rarray of the 'hydration' atomic radii.
These radii are used in construction of the solvent-accessible
(surface)
and can be found (and possibly redefined) in the
icm.hdt file.
- returns the Radius ( as charge ) rarray of the 'electrostatic' atomic radii.
These radii are used for building the skin (analytical molecular
surface) for electrostatic dielectric boundary calculation with
electroMethod = "boundary element".
These parameters can be found (and possibly redefined) in the icm.vwt file.
- returns a pseudo-random Random ( ) real in the range from 0. to 1.
- returns a pseudo-random Random ( i_max ) integer distributed in [1, i_max ]
- returns a pseudo-random Random ( i_min , i_max ) integer distributed in [ i_min , i_max ]
- returns a pseudo-random Random ( r_min , r_max ) real evenly distributed in [ r_min , r_max ]
- returns a Random ( r_min , r_max , i_n ) rarray [1: i_n ] with pseudo-random real values
distributed in [ r_min , r_max ]
- returns a Random ( r_mean , r_std , i_n , "gauss" ) rarray of i_n elements with normally distributed pseudo-random
values. The mean and standard deviation are provided as the first two arguments
- returns a Random ( r_min , r_max , i_nRows , i_nColumns ) matrix [1: i_nRows, 1: i_nColumns] with pseudo-random real values
distributed in [ r_min , r_max]
- returns a Random ( i_nRows, i_nColumns, r_min, r_max ) matrix [1: i_nRows, 1: i_nColumns] with pseudo-random real values
distributed in [ r_min, r_max ]
Examples:
print Random(5) # one of the following: 1 2 3 4 or 5
print Random(2,5) # one of the following: 2 3 4 or 5
print Random(2.,5.) # random real in [2.,5.]
randVec=Random(-1.,1.,3) # random 3-vector with components in [-1. 1.]
randVec=Random(3,-1.,1.) # the same as the previous command
randMat=Random(-1.,1.,3,3) # random 3x3 matrix with components in [-1. 1.]
randMat=Random(3,3,-1.,1.) # the same as the previous command
Random(0., 1., 10, "gauss" ) # normal distribution
- returns an Random( I_lengths s_alphabet )sarray of random strings of lengths specified in the I_lengths array.
Strings are comprised from the characters specified in the s_alphabet. Alphabet specifications are the same as
character set specifications in regular expressions: "A-Z", "\\w", "\\dA-Fa-f", "ACGT".
- returns an i_n element Random( i_n S_words ) sarray consisting of the words specified in the S_words array repeated in random order.
Random( Iarray(10,20) "\\dA-Z" ) # returns 10-element sarray with 20-character strings containing random digits and capital letters
Random( 100, {"rock","paper","scissors"} ) # returns 100-element sarray consisting of words "rock", "paper" and "scissors" in random order
Random( 100, Random( Iarray(10,3) "a-z" ) ) # returns 100-element sarray containing 10 random words from the "a-z" alphabet
- returns a Rarray ( i_NofElements ) rarray; creates zero-initialized rarray [1: i_NofElements].
You can also create an zero-size real array: Rarray(0) .
- returns a Rarray ( i_NofElements, r_Value ) rarray [1: i_NofElements ] with all elements set to r_Value.
- returns a Rarray ( i_NofElements, r_From, r_To ) rarray [1: i_NofElements ] with elements ranging from r_From to r_To.
- returns Rarray ( r_From, r_To , r_step ) rarray of equally spaced numbers from r_From to r_To. Example:
Rarray( 3.1, 15. 0.1)
- converts iarray into a Rarray ( iarray ) rarray.
- converts sarray into a Rarray ( sarray ) rarray.
- converts sarray into a Rarray ( sarray s_patternForValue1 ) rarray of 1. and 0. The value is 1. if an element if an array matches the string. E.g. Rarray({"M","W","M","E","W"},"M") # returns {1. 0. 1. 0. 0.}
- rounds the input array to the specified number of significant digits. If n is out of bounds ( less than zero or more than 12, the function switches to the default of 2.
Rarray ( R n_significantDigits ) → R_rounded
- extracts different groups of elements of the matrix and casts them into a Rarray ( M [ i_flag ] ) rarray.
There are seven (7) possibilities for a matrix n rows by m columns:
Examples:
a=Rarray(54) # create 54-th dimensional vector of zeros
a=Rarray(3,-1.) # create vector {-1.,-1.,-1.}
a=Rarray(5,1.,3.) # create vector {1., 1.5, 2., 2.5, 3.}
a=Rarray({1 2 3}) # create vector {1. 2. 3.}
a=Rarray({"1.5" "2" "-3.91"}) # create vector {1.5, 2., -3.91}
#
M=Matrix(2);M[2,2]=2;M[1,2]=3
Rarray( M )
Rarray( M 1 )
Rarray( M 2 )
Rarray( M 3 )
Rarray( M 4 )
Rarray( M 5 )
Rarray( M 6 )
Rarray( M 7 )
Rarray ( R_ali ali_from { seq | i_seqNumber } )
- returns a projected rarray. The R_ali rarray contains values
defined for each position of alignment ali_from. The function squeezes out the
values which correspond to insertions into sequence seq_, that is, in effect,
projects the alignment array R_ali onto sequence seq_.
E.g. for the residue conservation: Rarray( Rarray( alig ), alig , myseq ) -
Projecting from one sequence to another sequence via alignment.
Let us imagine that we have two sequences, seq1 and seq2 which take
part in multiple sequence alignment ali .
The transfer of property R1 from seq1 to seq2 can be achieved via two transfers :
Now R2 has the same dimension as seq2. Values aligned with seq1 are transferred by alignment,
other values are set to r_gapValues .
See also:
String( s_,R_,ali_,seq_ )function to project strings
Rarray( R_seq seq_ ali_to r_gapDefault )function to project from sequence to alignment
Probability()function
Rarray ( rarray reverse )
- converts input real array into an rarray with the reversed order of elements.
Example:
Rarray({1. 2. 3.} reverse) # returns {3. 2. 1.}
Rarray ( R_seq { seq | i_seqNumber } ali_to r_gapDefault )
- projects the input rarray from seq_ to ali_to (the previous function does it in the opposite direction).
The R_seq rarray contains values defined for each position of the sequence seq_.
The function fills the gap positions in the output array with the r_gapDefault values.
Combination of this and the previous functions allow you to project any numerical property of
one sequence to another by projecting the r1 property of seq1 first to the alignment and than back to
seq2 (e.g. Rarray( Rarray(r1,seq1,a,99.) , a, seq2) ).
This function can also be used to determine alignment index corresponding to a sequence index.
Example:
read alignment s_icmhome+"sh3"
t = Table(sh3)
group table t Count(Nof(t)) "n" append # add a column with 1,2,3,..
show t # t looks like this:
#>T t
#>-cons--------Fyn---------Spec--------Eps8-------n---
" " 0 1 1 1
" " 0 2 2 2
" " 0 3 3 3
" " 0 4 4 4
. 1 5 5 5
...
t2forFyn = t.Fyn == 2 # table row for position 2 in seq. Fyn
t2forFyn.n # corresponding alignment position
See also the
String( s_,R_,seq_,ali_,s_defChar ) function to project strings.
Rarray ( sequence R_26resProperty )
- returns a rarray of residue properties as defined by R_26resProperty for 26
residue types (all characters of the alphabet) and assigned according to the
amino-acid sequence.
Example with a hydrophobicity property vector:
s= Sequence("TTCCPSIVARSNFNVCRLPGTPEAICATYTGCIIIPGATCPGDYAN") # crambin sequence
# 26-dim. hydrophobicity vector for A,B,C,D,E,F,..
h={1.8,0.,2.5,-3.5,-3.5,2.8,-.4,-3.2,4.5,0.,-3.9,3.8,1.9,-3.5,.0,-1.6,-3.5,-4.5,-.8,-.7,0.,4.2,-.9,0.,-1.3,0.}
hs=Rarray(s,h) # h-array for each sequence position
hh = Smooth(Rarray(s,h), 5) # window average
Rarray ( ali [ simple|exact ] )
- returns a rarray of conservation values estimated as
mean pairwise scores for each position of a pairwise or multiple alignment.
The number is calculated as the sum of RESIDUE_COMPARISON_VALUES over n*(n-1)/2 pairs in
each column. The gapped parts of an alignment are considered equivalent to the 'X' residues
and the comparison values are taken from appropriate columns.
Option exact uses raw residue substitution values as defined in the comparison matrix
These values can be larger than one for the residues the conservation of which is important (e.g.
W to W match can be around 3. while A to A match is only about 0.5.
By default (no keyword) the matrix is normalized so that two identical residues contribute
the replacement value of 1. and two different but property-similar amino acid contributed values from 0. to
1. depending on the residue similarity.
If option simple is specified, each pair with identical residues contributes 1. while
each pair of different amino-acids contributes 0.
The resulting total of pairwise similarities d calculated for n(n-1)/2 pairs is then
converted into the conservation value with the following equation:
c = ½(1+ √(8·d +1) ) (a solution of the equation n2 - n = 2d ) and further divided by the number of sequences in the alignment
simple option and the default are therefore between 1/n (all residues are different) and 1.
To project the resulting array to a specific sequence, use the Rarray( R_ ali_ seq_ ) function (see above).
To calculate conservation with respect to a particular set of residues in
a structure, use the Score( rs_ [ simple ] ) function.
Example:
read alignment s_icmhome+"sh3"
show Rarray(sh3)
#
a=Rarray(sh3 simple) # a number for each alignment position
# to project a to a particular sequence, do the following
b=Rarray(a,sh3,Spec) # a number for each Spec residue
String(Rarray(a, sh3, Spec ))//String(Spec) # example
Score( ali [simple]) Entropy( ali [simple|info])
- converts integer to Real ( integer ) real number.
- converts string to Real ( string ) real number. The conversion routine ignores trailing non-numerical characters.
Examples:
See also: s = "5.3"
a = Real(s) # a = 5.3
s = "5.3abc" # will ignore 'abc'
a = Real(s) # the same, a = 5.3Toreal
Mod function.
function description example
Remainder(x,y) brings x to [-y/2 , y/2] range Remainder(17.,10.) → -3.
Mod(x,y) brings x to [0 , y] range Mod(17.,10.) → 7.
- returns the Remainder ( i_divisor, [ i_divider ] ) integer
- returns the Remainder ( r_divisor, [ r_divider ] ) real remainder r = x - n*y where n is the integer nearest the exact value of
x/y; if | n-x/y|=0.5 then n is even. r belongs to [ -|y|/2, |y|/2 ] range
- returns the Remainder ( iarray, [ i_divider ] ) iarray of remainders (see the previous definition).
- returns the Remainder ( rarray, [ r_divider ] ) rarray of remainders.
The default divider is 360. (real) or 360 (integer) since we mostly deal with angles.
Examples:
read object s_icmhome+"crn.ob"
# transform angle to the standard
# [-180., 180.] range. (Period=360 is implied)
phi=Remainder(Value(v_//phi))
# we assume that you have two objects
# with different conf. of the same molecule
Reference( seq [ s_fieldName ] )
(regular expressions, and case-sensitivity, - see below).
Replace( s|S s_regexp s_by regexp [i_field=0] ) → s|S
- returns a Replace ( s_source s_icmWildcard s_replacement ) string, which is a copy of the source string with globally substituted substrings
matching s_icmWildcard by the replacement string s_replacement.
Example:
a=Replace(" 1crn "," ","") # remove empty space
- make several replacements in a row. The size of the two arrays must be the same.
Replace ( s_source S_fromArray S_toArray )
Example which generates a complimentary DNA strand (actually there is
a special function Sequence( seq_, reverse ) which does it properly).
invertedSeq = String(0,1,"GTAAAGGGGTTTTCC") # result: CCTTT..
complSeq=Replace(invertedSeq,{"A","C","G","T"},{"T","G","C","A"})
# result: GGAAA...
- replace several strings by a single other string. If s_replacement is
empty, the found substrings will be deleted.
Replace ( s_source S_fromArray s_replacement )
Example which generates a complimentary DNA strand:
cleanStr=Replace("XXTEXTYYTEXT",{"XX","YY"},"")
- returns a Replace ( S s_icmWildcard s_replacement ) sarray with globally substituted elements (the original sarray remains intact).
Examples:
aa={"Terra" "Tera" "Teera" "Ttera"}
show column aa Replace(aa "er?" "ERR") Replace(aa "*[tT]" "Shm")
Replace ( S S_fromArray S_toArray ) or Replace ( S k_translation_name-val_collection [s_nonMatchFormat])) sarray with multiple substitutions.
- returns a Replace ( S s_icmWildcard s_replacement ) sarray with multiple substitutions to a single string.
Replace( S s_completeString s_by exact ) → S"never again" element of S will only be matched with the "never againg"
string, but not with "never" .
Replace( s|S s_whatAsIs s_byAsIs simple ) → s_|Ss="a[b]()c"
Replace(s,"[b]","()$",simple) # no intepretation Replace( s|S s_regexp s_by regexp [i_field = 0] ) → s|SReplace("bla 1"//"Bla 2"//"BLA 3" , "(?i)bla ","") # get rid of blaread string "t.html"
s_out = Replace( s_out, "(?n)<i>(.*?)</i>", "<b>\\1</b>" regexp ) # replace italic with bold
s_out = Replace( s_out, " +", " ",regexp ) # replace multiple spaces with a single on
","\n", exact)
S = Replace( S, "read table html "http://pfam.sanger.ac.uk/search/keyword?query=sh2" name = "sh2t"
sh2t.ID = Replace(sh2t.ID, "<.*?>","",regexp) Replace( chem , s_smartFROM, s_smileTO [exact] )exact option will supress the redrawing if the number of atoms in the FROM and TO
patterns is the same.
read table mol "drugs.sdf"
add column tt Replace(drugs.mol, "[R1]C(=O)O","[R1]C(=O)OC")
will modify in place. This replacement can be done only for the "terminal" fragments (one attachment point)
modify chemarray s_pattern s_repl [exact]
will iteratively delete selected atom pattern, e.g. "[*;D1]"
`Trim-chemical{Trim} ( X [s_smart] ...)
Res ( { os | ms | rs | as } [ append ] ) Res ( { rs [ append ] )
- selects residue(s) related to the specified objects ( os_), molecules ( ms_)
or atoms ( as_), respectively. Option append extends the selection with
the terminal residues (like Nter and Cter in peptides)
Examples:
show Res( Sphere(a_1/1/* 4.) ) # show residues within 4 A
# vicinity from the firsts one
See also: Atom ( ), Mol ( ), Obj ( ).
- returns Res ( ali { seq | i_sequence } ) residue selection corresponding to the aligned positions of the specified sequence.
The sequence can be specified by its order number in the alignment (e.g. 1crn_m in the example
below has number 1 ), or by name.
- returns the Resolution ( ) real resolution of the current object.
- returns the Resolution ( os_object ) rarray X-ray resolutions for the specified objects.
The resolution is taken from the PDB files.
Examples:
sort object Resolution(a_*.) # resort objects by resolution
res=Resolution(a_1crn.)[1]
print "PDB structure 1crn: resolution = ", res, " A"
- returns the Resolution ( s_pdbFileName pdb ) real resolution of the specified pdb-file. The function returns 9.90 if resolution is not found.
- returns the Resolution ( T_factors [ R_6cell ] ) rarray of X-ray resolution for each reflection of the specified structure factor table.
The resolution is calculated from h, k, l and cell parameters taken from R_6cell or the standard defCell shell rarray.
Example:
read factor "igd" # read h,k,l,fo table from a file
read pdb "1igd" # cell is defined there
defCell = Cell(a_) # extract the cell parameters from the object
group table append igd Resolution(igd) "res"
show igdset resolution
- returns Ring( as )logical yes all atoms from the selection belong to one ring
- returns subset of input Ring( vs ) variable selection which belongs to one ring
- returns chemical array of ring system(s)
Ring( chemical )
- returns chemical array of the smallest set of smallest rings (SSSR)
Example:
Ring( chemical simple ) show Smiles( Ring( Chemical("C(=CC=CC1C(=CC=C(C2C3)C=CC=3)C=2)C=1" ) ) unique )
show Smiles( Ring( Chemical("C(=CC=CC1C(=CC=C(C2C3)C=CC=3)C=2)C=1" ) simple ) unique )
- returns the Rfactor ( T_factors ) real R-factor residual calculated from the factor-table
elements T_factors.fo and T_factors.fc. Reflections marked with T_factors.free = 1 are ignored.
- returns the Rfree ( T_factors ) real R-factor residual calculated from the factor-table
elements T_factors.fo and T_factors.fc. Only reflections marked with T_factors.free = 1 will be used.
Rmsd ( { iarray | rarray | matrix | map } )
- returns the real standard deviation estimate (sigma) from the estimated mean for specified sets of numbers. This function returns the unbiased estimation of standard deviation with Bessel's correction according to this formula: s = √(∑(xᵢ-μ)²/(n-1) ), where μ is the sample mean.
Rmsd ( Rn Wn ) real weighted rmsd and weighted mean as r_out according to this formula:
xw = Sum(w[i]*x[i])/Sum(w[i]) # the weighted mean
np # is the number of non-zero weights
sdw2 = Sum(w[i]*(x[i]-xw)^2) / (((np-1)/np)*Sum(w[i]))
sdw = Sqrt(sdw2)
Rmsd ( as_tetheredAtoms ) returns the from the atoms to which they are real root-mean-square-deviation of selected atoms tethered . The distances
are calculated after optimal superposition according to the
equivalences derived from tethers (compare with the Srmsd( as_ ) function
which does not perform superposition ).
This function also returns the transformation in the R_out array.
Rmsd ( ms_select1 ms_select2 chemical [output] ) real root-mean-square distance between two selected chemical (hetero) molecules
after an optimal chemical superposition via graph-matching is performed.
In this mode atom equivalence can be found
either by substructure search or (if none of molecules is substructure of other) by common substructure search algorithm.
Other feature of chemical mode is that it enumerates topologically equivalent atoms to find best superposition.
The maximal common substructure will be used for the calculation.
Option output will produce R_2out array with individual deviation for the matched pairs. Rmsd(R_2out) will
essentially be the overall Rmsd, but one will be able to measure the maximal and median deviation as well.
Srmsd( ms1 ms2 chemical ) and superimpose command.
Rmsd ( chemarray ms_select2 [pharmacophore] ) Rmsd ( as_pharmTemplate as_select2 pharmacophore ) real root-mean-square distance between pharmacophore points of as_pharmTemplate and as_select2after an optimal superposition of as_pharmTemplatefind pharmacophore , show pharmacophore type , makePharma
Rmsd ( as_select1 as_select2 [ { { ali|align } | exact } ] )
- returns the real root-mean-square-distance between two aligned sets
after these two sets are optimally superimposed using McLachlan's algorithm.
Virtual atoms. Be default, the first two virtual atoms ( vt1 and vt2 ) are automatically
excluded from both selections unless the virtual option is explicitly specified.
The optional third argument defines how atom-atom alignment is
established between two selections (which can actually be of any level
atom selection `as_ , residue selection `rs_ , molecular selection `ms_ , or
object selection `os_ , see alignment options).
Number of equivalent atom pairs is saved in i_out .
Two output selections as_out and as2_out contain corresponding sets of equivalent atoms.
This function also returns the transformation in the R_out array.
See also: superimpose and Srmsd ().
Examples:
read pdb "1mbn" # load myoglobin
read pdb "1pbx" # load alpha and beta
# subunits of hemoglobin
print Rmsd(a_1.1 a_2.1 align) # myo- versus alpha subunit
# of hemo- all atoms
print Rmsd(a_1.1//ca a_2.1//ca align) # myo- versus alpha subunit
# of hemo- Ca-atoms
print Rmsd(a_1./4,29/ca a_2.1/2,102/cb exact) # exact match
- extracts the 3x3 rotation Rot ( R_12transformVector ) matrix from the transformation vector.
- returns Rot ( R_axis , r_Angle ) matrix of rotation around 3-dimensional real vector R_axis by angle r_Angle.
To solve the inverse problem, i.e. calculate the angle from a transformation, use the
Axis( R_12 ) function which returns the angle as r_out .
Examples:
# rotate molecule by 30 deg. around z-axis
rotate a_* Rot({0. 0. 1.},30.)
Rot ( R_3pivotPoint R_3axis , r_Angle )
- returns rarray of transformation vector
of rotation around 3-dimensional real vector R_axis by angle
r_Angle so that the pivotal point with coordinates R_3pivotPoint remains static.
Examples:
# rotate by 30 deg. around {0.,1.,0.} axis through the center of mass
nice "1crn"
R_pivot = Mean(Xyz(a_//*))
transform a_* Rot(R_pivot,{0. 1. 0.}, 30.)
- returns empty Sarray ( integer ) sarray of specified dimension
- returns Sarray ( integer s_Value ) sarray of specified dimension initialized with s_Value
- returns Sarray ( integer S_ids ) sarray of unique strings using the ID seeds. Examples:
Sarray(10,Sarray(0)) # ID1 ID2 etc.
Sarray(10,{"A"})
Sarray(10,{"A","B","C"})
#
read csv header name='b' s_icmhome + "bnames.csv" # 2K baby names
Sarray(10000,Shuffle(Unique(b.name,sort))) # useful for generating identifiers in tables
- returns Sarray ( s_wildCard directory [simple|all] ) sarray of file names with full path to them. With 'simple' option only file names are stored in the result array.
all toggles recursive search in sub-folders.
Sarray( "*.pdb" directory )
Sarray( "/home/user/*.ent*" directory )
if (Nof( Sarray("*.png") )==0) print "No images found"
- converts the input string into a ONE-dimensional Sarray ( string ) sarray .
To split a string into individual lines, or to split a string into a sarray of characters,
use the Split() function.
- converts input arrays into an Sarray ( iarray|rarray|sarray ) sarray.
- generates 32 char or 26 char MD5 based has string.
Example in which we create a unique chemical id:
Sarray ( sarray [32] hash ) add column t Chemical({"C1CCCC1","CCO"})
add column t Sarray(Smiles( t.mol unique cistrans ), 32 hash)
- converts input residue selection into an Sarray ( rs [ { append | name | residue } ]) sarray of residue ranges, e.g.:
{"a_a.b/2:5", "a_a.b/10:15",..} or with option residue into an array of individual selections (see also Name( rs full ) .
Options:
append : will merge residue ranges
name : will return a string array of residueName residueNumber records
residue : will return an array of selection strings a_obj.mol/residueNumber records
Example:
Sarray(a_/2,4:5 name)
#>S string_array
def.a1/ala2
def.a1/trp4
def.a1/glu5
Sarray(a_/2,4:5 residue)
#>S string_array
def.a1/2
def.a1/4
def.a1/5
Field(Sarray(a_/2,4:5 name),2,"/") # extract residues
#>S string_array
ala2
trp4
glu5
The l_showResCodeInSelection system logical controls if one-letter residue code is printed
in front of the residue number ( e.g. a_/^F23 instead of a_/23 ).
See also:
Name( rs full ) : preferred way to generate arrays of residue selection strings
String( rs_ ) which returns one string;
Label( rs_ | as_ ) which will format the output string according to the resLabelStyle or atomLabelStyle preference.
l_showResCodeInSelection
- creates a string representation of all the conformations in the Sarray ( stack, vs_var ) stack
Variable selection allows one to choose the conformational feature you want.
Character code:
Backbone (phi,psi pairs):
Sidechain (chi1):
Example:
(Note use of special PSI torsion in the last example.)
show Sarray(stack,v_/2:10/x*) # coding of side-chain conformations
show Sarray(stack,v_//phi,psi) # backbone conformation character coding
show Sarray(stack,v_/2:10/phi,PSI) # character coding of a chain fragment
Other examples:
ss=Sarray(5) # create empty sarray of 5 elements
ss[2]="thoughts" # assign string to the second element of the sarray
sa=Sarray("the first element")
show Sarray(Count(1 100)) # string array of numbers from 1 to 100
- Reversing the order of elements in an sarray
Sarray (sarray reverse )
- converts input sarray into an sarray with the reversed order of elements.
Example:
Sarray({"one","two"} reverse) # returns {"two","one"}
See also: Iarray( I_ reverse ), Rarray( S_ reverse ), String(0,1,s)
Sarray ( sarray i_from i_to )
returns sarray of substrings from position i_from to position i_to .
If i_from is greater than i_to the direction of substrings is reversed.
Example:
a={"123","12345"}
Sarray(a,2,3)
{"23","23"}
Sarray(a,5,2)
{"32","5432"}
- returns Sarray ( T_index ) sarray of index table elements
Example:
See also: read index "myindex"
S = myindex[2:8]
S[1]write index
Score( <R_X> <R_Y> ) => r [-1.:1.] # overlap between two distributions
Score( <R_Ei> <R_Di> <wE> <wD> ) => <r> # prediction quality
Score( <I_keys1> <I_keys2> <nBits>|<R_bitWeights> [simple] ) => M # Tanimoto distances
Score( <seq1> <seq2> [new|nucleotide|simple] ) => <R_scores>
Score( <seqArray[n]> ) => Mnn_alignedScore
Score( <i_len> <r_probability> [comp_matrix|similarity|identity] ) => <r_>
Score( <ali2> [area|sort|comp_matrix|similarity|identity [<i_alnLength>] ] ) => r # see also Distance (<ali>), and Rarray(<ali>)
Score( <X_n> <X_m> [[<R_Wn> <R_Wm>] <r_minScore> (0.4) [<r_steepness>(6.)]] set ) => r_inter_set_score [0:1]
Score( <X_3D_n> [<X_3D_m>] field|similarity|distance ) => <M_nxm APF_scores> # needs Chemical(<as> exact hydrogen)
Score( <X_3D_n> torsion ) => <R_Free_Strain> #
Score( <rs_n> [simple|info|comp_matrix] ) => <R_n_conserv_scores> # info is entropy
Score( <rs_n> <seq_n> ) => <r_score_without_alignment>
Score( <rs_n> <seq_n> all ) => <T_sel_scores_seqids>
Score( <model> full|test [<s_stats>] ) => <r_learnStatistics>
Score( predict <R|I_obs> <R|I_pred> [<R_weights>] ) => <R_allRegression_or_Classification_Stats>
Score ( R_1, R_2 ) → r_overlapMeasurereal measure of overlap between two real arrays.
This measure varies between -1 and 1..
(all values of R_1 are smaller than all values of R_2) and +1.
(all values of R_1 are greater than all values of R_2)
and may serve as a ranking criterion.
Examples:
show Score({1. 2. 5. 3.} {3. 1.5 1.5 5.}) # 0. perfectly overlapping arrays
show Score({2. 5. 3.} {1. 1.5 0.5}) # 1. no overlap R_1 > R_2
show Score({1. 1.5 0.5} {2. 5. 3.}) # -1. no overlap R_2 > R_1
show 1.-Abs(Score({1. 3. 2.5} {2. 5. 3.})) # relative overlap between R1 and R2 Score( X_n X_m [[R_Wn R_Wm] r_minScore (0.3) [r_steepness(6.)]] set ) → r_sim_score [0:1]
Other returned values:
Example:
r_out : N11 r_2out : N22 r_3out : N12 Score( t.mol tt.mol set ) Score( X_3Dn torsion ) → R_n_free_strain Score( X_3Dn [X_3Dm] field|similarity|distance ) → M_nxm apf_scores
An array can be generated with read pmf s_icmhome + "APF"Chemical ( ms exact ) function.
If one array is provided an n by n matrix with all pairwise scores is calculated.
The options:
field : returns raw (negative for similar atom pairs) pairwise APF similarity scores Sij
similarity : returns normalized APF similarity calculated as S_ij_sim = |Sij|/Sqrt(|Sii*Sij|).
(we change the signs on Sij values to make it positive if necessary).
The similarities are 1. for self-comparison and numbers less for different molecules.
distance : returns chemical distance calculated as 1. - normalized_similarity .
build string "H"
build string "W"
build string "A"
add column t Chemical(a_*. exact hydrogen ) name="mol"
read pmf s_icmhome+"APF"
show Score(a_1. a_2. field)
sf = Score( t.mol field )
ss = Score( t.mol similarity )
sd = Score( t.mol distance )
CS = Rarray( ss 6 ) # off diagonal elements
Mean(CS) # average similarityexact
macro which makes the superposition and returns the un-normalized score. The normalized score can be returned
after converting the superimposed selections into a 3D chemical array with the Chemical( as exact ) function
Alternatively, the normalization can be done directly by the above formula ( S_ij = |Sij|/Sqrt(|Sii*Sij|) if self-scores are calculated.
Score ( model [ test | full ] s_stats ) → r_PredictionQuality
Note that only measure abbreviation/formula description
"tpos" TP number of true positives
"fneg" FN number of false negatives
"fpos" FP number of false positivers
"tneg" TN number of true negatives
"accuracy" Q=(TP+TN)/(TP+TN+FP+FN) fraction of correctly predicted label assignments
"sensitivity" TP/(TP+FN) fraction of correction predicted positive labels
"specificity" TN/(TN+FP) fraction of correction predicted negative labels
"mathews" (TP*TN-FN*FP)/Sqrt((TP+FN)(TP+FP)(TN+FN)(TN+FP)) Mathews correlation
"precision" PR=TP/(TP+FP)
"recall" RE=TP/(TN+FP) same as sensitivity
"f1" 2*PR*RE/(PR+RE)
"acuracy", and "mathews" are overall measures symmetrical with respect
to the label. Frequently a method is characterized by an area under a recall - precision curve.
measure formula description
"r2" r=Mean((X- correlation squared
"rmse" Sqrt(Sum((Xpred-Xobs)^2 )/N) root-mean-squared error
"expavg"
Score ( R_En, R_Dn, wE, wD ) → r_PredictionQuality
Evaluates the quality of submitted multiple predictions for a unknown outcome.
The submitted provies R_En , the evaluator evaluates R_Dn from the correct answer, then plugs in the weights and calculates the quality.
The Q (quality)-value of predicted "energies" R_En for n - states, by comparing predicted energies with the deviations R_Dn from the correct answer.
In essence we are doing the following:
For example, if you predicted n conformations with energies
E[n] and calculated RMSD deviations D[n] for each of those conformations from the correct conformation, the
quality of your prediction will be calculated by this function.
exp( -w*D ) [0., 1.]
The Q-value is calculated as follows:
Q = - Log( Sum( exp(- wE#(Ei-Emin) -wD*Di )) / Sum(exp(- wE#(Ei-Emin))) )
The best Q -value is 0. (it means that zero deviation (Di=0.) correspond to the best energy and the energy gap is large.
The weighting factors wE and wD can be used to change the relative contributions of energies and deviations.
Score ( sequence1, sequence2 )
- returns the real score of the Needleman and Wunsch alignment.
Each pair of aligned residues contributes according to the current
residue comparison table,
which is normalized so that the average diagonal element is 1.
Insertions and deletions reduce the score according to the gapOpen
and gapExtension parameters.
Approximately, the score is equal to the number of residue identities.
To calculate an array of mean scores for each column of a multiple sequence alignments use the Rarray( ali [ exact ] ) function.
i_out returns the number of identical residues.
Examples:
read sequence msf s_icmhome+"azurins.msf"
a = Score( Azur_Alcde Azur_Alcfa ) # it is around 90. Score ( seq_n_long, seq_m_short simple ) → R_n-m+1_scores Score ( seq_n, rs_N ) → r_no_gap_score Score ( seq_n, rs_N all ) → T_N-n_scores_ids_for_all_framesall option.
i (relative number), nu (first residue number), sl (fragment selection string), se (the first residue code), sc (normalized alignment score divided by the sequence selfscore and multiplied by 100., id (sequence identity), sf (relative surface area), ss ( relative non-loopsecondary structure ). id , sf , ss range from 0. to 100. % .
Make sure to assign the secondary structure and calculate the atomic surface areas before you fun the Score(.. all ) function.
Example:
build string "ASDFY"
a=Sequence("SDF")
assign sstructure a_/A
show surface area
t = Score( a_/A a all)
show tDistance( ).
Score ( rs, [ simple | comp_matrix | info ] ) Rarray( ali )
The function returns the rarray of alignment-derived conservation values for the selected residues.
For each residue Ri in the residue selection rs_ the following steps are taken:
The default mode shows positions with residues of the same type as more conserved than positions
with residues of different types.
simple mode: Cij = 1. for two identical residues and 0. otherwise . It is also the default.
comp_matrix (also the default) mode: Cij is taken from a normalized comp_matrix . Its elements are calculated as Cij_norm = Cij/Sqrt(Cii*Cjj) with the negative elements set to zero. The sum, d, of normalized positive similarity values cij is then converted to the conservation value of (1/2·nseq) √(8d+1) .
info (inverse information entropy mode): Cij = 1./(1.+Entropy), where Entropy= -Sum((fj)Log(fj)). The elements after normalization range from 0. (no conservation, 1. conservation).
Example in which we compare conservation on the surface and in the core:
read alignment s_icmhome+"sh3.ali"
read pdb "1fyn"
make sequence a_a
group sequence sh3
align sh3
display ribbon
color ribbon a_a/A Score( a_a/A simple )
show surface area
show Mean( Score( Acc(a_a/*) ) ) # conservation score for the surface
show Mean( Score( a_a & !Acc(a_/*)))# conservation score for the buriedRarray( ali [simple] )
Score ( ali2, [ { identity | similarity | comp_matrix | sort } [i_alnLength] )
- returns the real score of the given pairwise alignment calculated by different methods. By default the score (or number of identical pairs) are divided by the minimal length of the two sequences. With the third i_alnLength argument, it will be divided the this argument. If the 3rd argument is zero, the score/identity will be divided by the length of the alignment.
For a straitforward alignment conservation see: Rarray( ali [simple] )
Options:
Needleman and Wunsch score: aligned residues score according to the
residue comparison table, gaps according to the gapOpen and gapExtension parameters.
identity
the number of identical residues in the alignment divided by the smallest sequence length
and multiplied by 100 %.
comp_matrix
the alignment score without the gap component.
It contains only the total score of the aligned residues calculated from the
residue comparison table and does not include penalty term.
similarity
the alignment score without the gap component multiplied by 100. and divided by the smallest sequence length.
sort
a score occasionally used for ranking/sorting the alignments in fold recognition.
Currently it is equal to the comp_matrix_score - 1.3*totalGapPenalty
Align( ali I_seqIndexes ) function, e.g.
make sequence 5 20 # 5 random sequences of length 20
align sequence # creates aln
Score( Align(aln, 1//2 ), identity) # 1//2 results in an iarray {1,2}n = Nof(sequence)
mdist = (Matrix(n,n,1.) - Distance(Parray(sequence ) )) * 100. # 100 for identities
- returns the Score ( i_minLen, r_Probability [, { identity | similarity | sort } ] ) real threshold score at a given r_Probability level
of occurrence of alignment with a protein of unrelated fold. The threshold is related to
the corresponding method of the score calculation (see above).
For example,
gives you the sequence identity percentage for sequences of
150 residues at which only one false positive is expected in a search through the
Swissprot database of 55000 sequences.
Score( 150, 1./55000.,identity)
See also the inverse function: Probability .
Select() → as_graph_or_displayed_or_current Select(as_source cond ) → as # conditions: "c" from x,y,z,b,o,c,f,a,u,v,w,n e.g. Select(a_ "b>80") Select(os|ms|rs|as s_expr|fieldName|cond ) → o|m|r|as # 'n' number_of, 'r' resolution Select(os|ms "biomt" i_Biomol ) → ms_of_biomol_i # molecules selected in i_Biomol Select(as_source I_indices ) → as Select(as_source hydrogen|hbond|smooth ) → as_expandByTerminalAtoms Select(as_source fix|unfix) → as_atomsOn(un)FixedBranches Select(as_source bond nNeighbors ) → as_atomsWithN_neighbors Select(as_source molecule i_mol ) → as_atoms_of_ith_disconnected_fragment Select(as grob ) → as_subset_in_grob_vicinity Select(grob ) → as_currentObjAtomsNearGrob Select(rs_patches i_MaxGapSize ) → rs_patchSmallGaps # e.g. Select(a_/1:2,4:6 2) → a_/1:6 Select(rs_patches margin,i_size ) → rs_expand_by_margin Select(as error ) → as_flankingBackboneBreak Select(as_inObjA os_objB ) → as_inObjB Select(seq [ ms_where [r_min_seqid(0.2) [r_mx_length_dist(0.3)]]] ) → ms_sim_seq
Select( as delete | error ) → as_bad_atom_pairsread pdb "2pe0"
display ribbon
display residue label Res(Select(a_ delete ))
select atoms of the fixed or rotatable branches, for the Select ( as fix | unfix ) fix or unfix options, respectively.
Select( as bond i_NofBondedAtoms )build smiles "CCO"
show Select(a_ bond 1) # selects all terminal hydrogens
show Select(a_ bond 2) # selects oxygen that is bonded to C and H
show Select(a_ bond 3) # no atom has three neighbors:
show Select(a_ bond 4) # carbons have 4 neighbors selecting by SMARTS patterns
Select ( os "n==nofMolecules" )
selects objects by number of molecules in them
Select ( ms "n==nofResidues" )
selects molecules by number of residues in them
Select( a_A,N "n==1" ) # all single residue amino or nucl molecules
Select( a_A,N "n>1" ) # all longer residue amino or nucl molecules
- returns either selected ( Select ( [ residue | molecule | object ] ) as_graph ) or displayed atoms ( a_*.//DD ).
By providing the argument, you can change the selection level.
Example:
display skin Select(residue)
- returns the source selection expanded to single covalently bonded atoms, e.g. hydrogens.
The returned selection is at the atomic level.
Options:
Select ( as_source [ hbond | hydrogen | smooth ] )
Example:
hydrogen : adds all attached hydrogens
hbond : adds all polar (non carbon connected, sorry, no aromatic hbonds) hydrogens
smooth : adds all bonded terminal atoms (hydrogens or heavy atoms).
Select(a_/tyr/o* hbond ) # adds hh to this selection
Select(a_/tyr/cb hydrogen ) # adds hb1 and hb2
Select(a_//ca,c,n smooth ) # carbonyl oxygen and N-terminal hydrogens
- returns a sub-selection of
Select ( as s_condition [ r_Value] ) atom selection as_ according to the specified condition s_condition.
- can use Select ( os|ms|rs s_condition ) field names (see set field sel name=.. ) or presets 'n' for number of molecules in an object
or number of residues in a molecule. Also 'r' for resolution (eg 'r<2.3' )
Three example conditions:
"X >= 2.0" , "Bfactor != 25." , "charge == 0." . Allowed properties and their aliases (case does not matter, the first character is sufficient) are as follows:
Note: do not forget to calculate surface in advance with the show area command.
Allowed comparisons: (== != > < >= <=).
The value can either be specified inside the string or used as a separate argument r_Value.
Field function.
See also the related functions:
Area, Bfactor, Xyz, Charge, Field .
Examples:
build string "se glu arg"
show Select(a_//* "charge < 0.")|Select(a_//c* "x> -2.4")
show Select(a_//c* , "x>", -2.4)
show Select(a_/* , "w>3.") # 3rd res. user field greater than 3.
Note:
atoms with certain Cartesian coordinates can also be selected by multiplying selection to a box
specified by 6 real numbers {x,y,z,X,Y,Z}, e.g.
show a_//* & {-1.,10.,2.,25.,30.,22.} or
a_//* & Box( ).
See also: display box and the Box function.
Select ( as_sourceSelection os_targetObject )
- returns the source selection as_sourceSelection from a source object which is
transferred to another object ( os_targetObject ).
The two objects must be identical in content. Example:
build string "ASD"
aa = a_/2/c* # selection in the current obj a_
copy a_ "b" # a copy of the source object
bb = Select(aa,a_b.) # selection aa moved to a_b.
Select ( os_sourceObject S_residueSelStrings ) Sarray ( rs_ residue ) function. The object name can be skipped. E.g.
Select( a_2ins. {"a/14","b/14"} ) Select ( os_sourceObject I_atomNumbers )
- returns atom selection of relative atom numbers in specified object os_sourceObject.
The iarray can be generated with the Iarray ( as_ ) function.
This function allows one to pass selections between ICM sessions.
Select ( rs_fragmented_selection [smooth|margin] i_gapSizeToHeal ) smooth) will take a source residue selection,
identify all gaps of size below the specified parameter and
will healing those gaps by adding them to the selection.
Select(a_/1,2,5,6 2 ) # residues 3 and 4 will be added
a_/1,2,3,4,5,6margin will simply expand the source selection by the specified margin size.
Select ( as_source "vw,14,hb,el,cn,tz" ) show energy command has
been used at list once. See example below.
tethers ("tz") this function
returns a selection of the static destination atoms (same as a_//Z ).
Example:
build string "se ala his trp"
copy a_ "tz" tether # make a copy object and tether atoms to a_tz.
show energy "vw,hb"
aca = a_//ca # selection of Ca atoms
Select( aca "vw,hb" )
Obj(Select( aca "tz" ))
2 a_tz. Type: ICM Mol: 1 Res: 1 def Select(seq [ ms_where [r_min_seqid(0.2) [r_mx_length_dist(0.5)]]] ) → ms_sim_seq
Example:
Sequence(a_1.a)
read pdb "1crn"
read pdb "2ins"
Select( Sequence( a_2.2 ) )
display ribbon a_
color ribbon magenta Select( Sequence( a_2.2 ) a_*. 0.2 0.3 ) Select( rs_|as_ alig ) → selection_propagated_by_aliSphere function with a coordinate matrix argument.
We need to follow these steps:
See also Xyz(as_res) will return a set of coordinates (as a Nx3 matrix) for the selected residue atoms. Use as_res & a_*.//!h* for heavy atoms only.
Xyz( as residue ) .
read pdb "1crn"
display
center_of_mass = Mean(Xyz(a_/44))
display xstick magenta Res(Sphere( center_of_mass , a_1. & a_/!44 , 7.5) )
# Res is added to select all residue atoms once an atom is inside the sphereread pdb "1crn"
display
center_of_mass = Mean(Xyz(a_/44))
nb = Res(Sphere( center_of_mass , a_1crn. & a_1crn./!44 , 7.5) )
if(Nof(nb)>0) then
group table t Rarray(0) "dist" Sarray(0) "sel"
for j=1,Nof(nb)
add t
cmj = Mean(Xyz(nb[j]))
t.dist[j] = Distance( center_of_mass, cmj )
t.sel[j] = String(nb[j])
endfor
sort t.dist # the smallest distance is on top ([1]) now
s_closest_res = t.sel[1]
endifGroup function with the "mean" argument.
This solution can also be modified to use the closest atom (instead of the center of mass) by using "min".
- returns Sequence ( as_select ) sequence extracted from specified residues.
- converts a string (e.g. "ASDFTREW") into an ICM Sequence( s [ nucleotide | protein ] ) sequence object.
By default the type is "protein". To reset the type use the
set type seq { nucleotide | protein } command.
seqA = Sequence( a_1./15:89 ) # create sequence object
# with fragment 15:89
show Align(seq1, Sequence("HFGD--KLS AREWDDIPYQ")
# non-characters will be squeezed out
a=Sequence("ACTGGGA", nucleotide)
Type(a , 2) # returns the type-string :
nucleotide
- returns a parray of sequenceswhich from the Sequence ( ali ) alignment.
- returns a chimeric Sequence ( ali --group ) sequence which represents the strongest character in every
alignment position.
- returns a chimeric Sequence ( profile ) sequence which represents the strongest character in every
profile position.
- converts DNA sequence to RNA sequence or the other way around, and returns a sequenced with T replaced by U or the other way around, depending on the source sequence.
Sequence ( seq_NAseq convert ) dn = Sequence("AATTCCGG" nucleotide) #create a DNA sequence
Type(dn, 2)
dna
rn = Sequence( dn, convert ) # converting seq dn to RNA
Type(rn, 2)
rna
show dn, rn
>dn
AATTCCGG
>rn
AAUUCCGG
back_to_dna = Sequence( rn , convert )
- returns the reverse complement DNA of RNA sequence:
Sequence ( seq_NAsequence reverse ) nucleotide |complement
_____________________|__________
|
A = Adenosine | T (replace by U for RNA)
C = Cytidine | G
G = Guanosine | C
T = Thymidine | A
U = Uridine | A
R = puRine (G A) | Y
Y = pYrimidine(T C) | R
K = Keto (G T) | M
M = aMino (A C) | K
S = Strong (G C) | S
W = Weak (A T) | W
B = !A (G T C) | V
D = !C (G A T) | H
H = !G (A C T) | D
V = !T (G C A) | B
N = aNy | N
- converts Sequence( S_sequenceString [S_seqNames] )sarray of sequence strings to protein-sequence parray
- returns a Sequence( S_namesOfLoadedSequences name )parray of protein sequences retrieved by name from the ICM shell/workspace.
add column T Sequence( {"MILERR", "STAGKVIKCKAAVLW"} {"aa","bb"} ) name="seq"
add column T Length(T.seq) name="len"
set name T.seq {"seq1", "seq2"} # reset names
sq1 = T.seq[1]
read alignment s_icmhome + "sh3.ali"
add column t Sequence({"Spec","Fyn"} name)sequence parray.
Shuffle ( I | R | S ) → shuffled_array Shuffle ( string ) → shuffled_string Shuffle ( seq ) → shuffled_sequencea={1 2 3}
Shuffle(a) # {2 1 3}
Shuffle(a) # {3 1 2}
Shuffle("this") # won't tell you..
- returns Sign ( real ) real sign of the argument.
- returns Sign ( integer ) integer .
- returns Sign ( iarray ) iarray.
- returns Sign ( rarray ) rarray.
- returns Sign ( map ) map with -1., 0., 1. values.
Examples:
Sign(-23)
-1
Sign(-23.3)
-1.
Sign({-23,13})
{-1,1}
Sign({-23.0,13.1})
{-1.,1.}
- returns the Sin ({ real | integer } ) real sine of its real or integer argument.
- returns the Sin ( rarray ) rarray of sines of rarray elements.
Examples:
print Sin(90.) # equal to 1
print Sin(90) # the same
print Sin({-90., 0., 90.}) # returns {-1., 0., 1.}
- returns the Sinh ( { real | integer } ) real hyperbolic sine of its real or integer argument.
Sinh(x)=0.5( eiz - e-iz )
- returns the Sinh ( rarray ) rarray of hyperbolic sines of rarray elements.
Examples:
print Sinh(1.) # equal to 1.175201
print Sinh(1) # the same
print Sinh({-1., 0., 1.}) # returns {-1.175201, 0., 1.175201}
site selection function
- returns the Site ( s_siteID [ ms ]) iarray of the site numbers in the selected molecule.
The default is all the molecules of the current object.
Example:
nice "1est" # contains some sites
delete site a_1 Site("cat",a_1)
Slide( ) slide). Slides
include the following:
In addition slides may contain window layout information, sufficient to restore the view
which was used when the slide was created.
parray of a view type.
These "slides" can be written and read as parts of the .icb project files with the read binary command.
slide1 = slideshow.slides[1]
display slide1 Slide( gui )slide containing only the current window layout information.
String slide gui, add slide.
Smiles ( as ) Smiles ( chem [unique] [cistrans|cartesian] ) smiles - string with the text representation of
the chemical structure of a selected fragment or a chemical array.
unique option will make that string independent of the order atoms in the molecule.
The cartesian option will adds 2D or 3D coordinates at the end of the result smiles string. That coordinates will be used in Chemical function
read object s_icmhome + "biotin.ob"
s_smiles = Smiles( Chemical( a_ exact hydrogen ) cartesian ) # coordinates will be preserved
read mol input=String( Chemical( s_smiles )) #
Srmsd( a_1. a_2. chemical )
#
# or even simpler
#
s_sm3d = "CCCCC|3D:1.39,-0.00,-0.02,2.16,1.30,-0.01,3.45,1.20,-0.82,4.23,2.52,-0.81,5.51,2.42,-1.62"
read mol input=String(Chemical( s_sm3d ) name="cc"
See also: build smiles, String( as_ ) - chemical formula.
- returns the window-averaged Smooth ( R_source, [ i_windowSize ] ) rarray.
The array is of the same dimension as the R_source and
i_windowSize is set to windowSize
by default. An average value is assigned to the middle element
of the window. i_windowSize must be an odd number.
At the array boundaries the number of averaged elements is gradually reduced to one element,
i.e. if i_windowSize=5, the 3rd element of the smoothed array will get
the mean of R1,R2,R3,R4,R5, the second element will get the mean of R1,R2 and R3,
and the first element will be set to R1.
- returns the Smooth ( R_source, R_weightArray ) rarray of the same dimension as the R_source,
performs convolution of these two arrays. If R_weightArray contains
equal numbers of 1./ i_windowSize, it is equivalent to the
previous option. For averaging, elements of R_weightArray
are automatically normalized so that the sum of all elements in the
window is 1.0.
Normalization is not applied if the sum of elements
in the R_weightArray is zero. Convolution with such an array
may help you to get the derivatives of the R_source array. Use:
{-1.,1.}/Xstep # for the first derivative
{1.,-2.,1.}/(Xstep*Xstep) # for the second derivative
{-1.,3.,-3.,1.}/(Xstep*Xstep*Xstep) # for the third derivative
# ... etc.
Examples:
gauss=Exp( -Power(Rarray(31,-1.,1.) , 2) ) # N(0.,1.) distribution on a grid
x = Rarray(361,-180.,180.) # x-array grows from 0. to 180.
a = Sin(x) + Random(-0.1,0.1,361) # noisy sine
b = Smooth(a,gauss) # gauss averaging
# see how noise and smooth signals look
plot x//x a//b display {-180.,180.,30.,10.}
# take the first derivative of Sin(x)
c = Smooth(Sin(x),{-1., 1.}) * 180.0 / Pi
# plot the derivative
plot x c display {"X","d(Sin(X))/dX","Derivative"} Smooth ( M_source, [ i_halfwindow (1)> [<r_radius (1.)>]] ) → <Mi,j get averaged with the values in the neighboring [i-n:i+n] [j-n:j+n] cells
, (2n+1)^2 in total, according to the gaussian weights calculated as exp( r2 / R2 ), where R is the r_radius parameter,
and n is the i_windowSize parameter. The default parameters are 1 for the i_halfwindow (corresponding to 9 cell averaging)
and the radius of 1..
Smooth(Matrix(10),0) # keeps the matrix intact
Smooth(Matrix(10),3,1.5) # weighted average with 7*7 surrounding values (7=3*2+1) for each cell.
- Gaussian averaging of property array R_property of residues rs_ .
The averaging is performed according to the spatial distance between residue Ca atoms.
The function returns the Smooth ( rs, R_property, r_smoothRadius ) rarray of the residue property AVERAGED in 3D using spherical Gaussian with
sigma of r_smoothRadius. Each residue contributes to the smoothed property with
the weight of exp(-Dist_i_j2/ r_smoothRadius2).
The inter-residue distances Dist_i_j are calculated between atoms carrying the residue label
(normally a_//ca).
These atoms can be changed with the
set label command.
Array R_1 is normalized so that the mean value is not changed.
The distances are calculated between
Examples:
nice "1tet" # it is a macro displaying ribbon++
R = Bfactor(a_/A ) # an array we will be 3D-averaging
color ribbon a_/A Smooth(a_/A R 1.)//5.//30. # averaging with 1A radius
color ribbon a_/A Smooth(a_/A R 5.)//5.//30. # with 5A radius
color ribbon a_/A Smooth(a_/A R 10.)//5.//30. # with 10A radius
# 5.//30. are appended for color scaling from 5. (blue) to 30.(red)
# rather than automated rescaling to the current range
set field a_/A Smooth(a_/A R 5.)
show Select( a_/A "u>30." ) # select residues with 1st field > 30.
- returns a transformation of the initial Smooth ( ali, [ i_gapExpansionSize ] ) alignment in which every gap is widened by the
i_gapExpansionSize residues. This transformation is useful
in modeling by homology since the residue pairs flanking gaps usually
deviate from the template positions.
The default i_gapExpansionSize is 1 (the gaps are expanded by one residue)
Smooth ( map , [ "expand" ] )
weighted 3D-window averaging
- returns Smooth( map ) map with averaged map function values.
By default the value in each grid node is averaged
with the six immediate neighbors (analogous to one-dimensional averaging
by Smooth(R,{1.,2.,1.}) . By applying Smooth several times
you may effectively increase the window.
This operation may be applied to "ge","gb","gs" and electron density maps
low-values propagation
- returns Smooth( map "expand" ) map in which the low values were
propagated in three dimensions to the neighboring nodes. This trick
allows one to generate more permission van der Waals maps.
This operation may be applied to "gh","gc" and electron density maps.
Examples:
m_gc = Smooth(Smooth(m_gc "expand"), "expand" )
See also: map , GRID.gcghExteriorPenalty .
SolveQuadratic( r_a r_b r_c | R_3|2 [all] ) → R_rootsall : returns two complex numbers: {r1,i1,r2,i2}
Example:
rts = SolveQuadratic(1. 2. 1.) # one real root
show rts
#>R
1.
Nof(rts) # number of roots
1
SolveQuadratic(1. 2. 3. all) # two complex roots
#>R
1.
1.414214
1.
-1.414214SolveQubic
SolveCubic( {r_a r_b r_c r_d | R_[a]bcd} [all] ) → R_realRoots|R_6re,imall : returns three complex numbers: {r1,i1,r2,i2,r3,i3}
SolveQubic(1.,-3.,3.,-1.) # identical roots of 1.
SolveQubic(1.,3.,3.,1.,all) # three pairs of rootsSolveQuadratic
- returns the Sql ( connect s_host s_loginName s_password s_dbName ) logical status of connection to the specified server.
The arguments are the following:
"localhost") - the host name
"root")
- returns the Sql ( s_SQLquery ) table of the selected records.
Some SQL commands are not really queries and do not return records, but rather
perform certain operations (e.g. insert or update records, shows statistics).
In this case an empty table is returned.
An example:
if !Sql( connect "localhost","john","secret","swiss") print "Error"
id=24
T =Sql( "SELECT * FROM swissprothits WHERE featureid="+id )
sort T.featureid
web T
# Another example
tusers = Sql("select * from user where User=" + s_usrName )
Sql(off)
- disconnects from the database server and returns the Sql ( off ) logical status.
See also: molcart, query molcart
- returns the Sqrt ( real ) real square root of its real argument
- returns Sqrt ( rarray ) rarray of square roots of the rarray elements.
- returns Sqrt ( matrix ) matrix of square roots of the matrix elements.
Examples:
show Sqrt(4.) # 2.
show Sqrt({4. 6.25}) # {2. 2.5}
selectSphereRadius .
ICM-shell variable which is equal to 5.0 A by default.
Sphere (as_source| grob|R_xyz|M_xyz [as_whereToSelect] [radius(5.)] ) → selection Sphere (as_source|M_xyz as_whereToSelect radius object|molecule|residue ) → os|ml|re selection of atoms in a certain
vicinity of the following set of points:
Xyz function)
The function can be much accelerated if you specify the desired
level of the resulting selection
explicitly (e.g. molecule ,or residue, or object in the second function template). For example, if you just want to know
molecules around a selection, you can say Sphere( a_1 a_2 6. molecule )
Res , Mol , and Obj )
can also be used to convert the atom selection into residues, molecules or objects, respectively
(e.g. Res(Sphere(a_/15,4.)) ), if speed is not an issue or the explicit level option is not available.
show Sphere( a_subA/14:15/ca,c,n,o , 5.2)
Res(Sphere( a_1.2 a_2.)) # residues of a_2. around ligand a_1.2
Sphere( a_1.2 a_2. 7. residue) # same but much fasterSphere( a_//a1 a_//a2 , -1.2 ) specifies the van der Waals gap of 1.2 , i.e.
interatomic_distance / (R(a1) + R(a2)) will be compared with 1.2
read pdb "2ins"
Sphere(a_1.1 a_1.2//!h* 3.4 ) # the traditional method
Sphere(a_1.1 a_1.2//!h* , -1.1) # the corrected method Sphere (M_xyz as_whereToSelect radius ring ) → as_ring_atoms selection of atoms in rings which centers are in a certain
vicinity of the set of points in M_xyz.
read pdb "2l8h"
display xstick a_
make grob skin a_2l8h.a2 a_2l8h.a2 name="g_skin"
color g_skin white
color g_skin black distance Xyz( Sphere( Xyz(g_skin) a_N,A//RA 3.5 ring ) ring ) GROB.atomSphereRadius = 2.5
display solid smooth g_skin
printf " Info> %.2f%% of ligand surface is exposed to aromatic contact\n" (1-Mean(Mean(Color(g_skin))))*100
returns soapMessage object with specified method name.
SoapMessage( s_methodName s_methodNamespace )
adds a number of name/values pairs to the exiting soap message and returns
a new soap message as a result
SoapMessage( soapMessage [ s_argumentName argumentValue ] ... )
parses xml source and returns soapMessage object.
SoapMessage( s_xmlSource )# create a message with soap method 'doGoogleSearch'
req = SoapMessage( "doGoogleSearch","urn:GoogleSearch" )
# add method arguments
req = SoapMessage( req, "key","btnHoYxQFHKZvePMa/onfB2tXKBJisej" ) # get key from google
req = SoapMessage( req, "q", "molsoft" ) # search 'molsoft'
# some other mandatory arguments of 'doGoogleSearch'
req = SoapMessage( req, "start" 0, "maxResults" 10 )
req = SoapMessage( req, "filter", no, "restrict", "", "safeSearch", no )
req = SoapMessage( req, "lr", "", "ie" "latin1", "oe", "latin1" )
HTTP.postContentType = "text/xml"
read string "http://api.google.com/search/beta2" + " " + String(req)
# parse the result and check it for errors
res = SoapMessage( s_out )
if Error(res) != "" print "Soap error: ", Error(res) SOAP services for more information.
Sort ( sarray|iarray|rarray|chemarray [reverse] ) reverse toggles the sorting order.
Examples:
See also: count_unique=Nof(Sort(Unique({1, 11, 7, 2, 2, 7, 11, 1, 7}))) # counts unique elementsUnique( ), sort.
# to split into characters
*
Split( s s_sepChars ) → S_words Split( s s_sep exact|regexp ) → S_words
# "A:1 B:2 C:3.3" into columns A,B,C, inverse to Sum(t,{"t.A","t.B"} " " ":")
Split( S s_sepCols s_sepColNameValue [exact|regexp] ) → T_words Split( chem_1 [chiral|tautomer|mol|group] )→chem_multi # see also: enumerate ..
# needs: make cluster t
Split( table_n.cluster [r_threshold|i_nGroups] ) → I_n_groupIndices
- returns Split ( s_multiFieldString, s_Separators ) sarray of parts of the input string separated by s_Separators.
Examples:
See also: lines=Split("a 1 \n 2","\n") # returns 2-array of {"a 1" " 2 "}
flds =Split("a b c") # returns 3-array of {"a" "b" "c"}
flds =Split("a b:::c",":") # returns 4-array of {"a b","","","c"}
resi =Split("ACDFTYRWAS","") # splits into individual characters
# {"A","C","D","F",...}Field( ).
- returns Split ( s_multiFieldString s_separator exact ) sarray
of fields separated exactly by s_separator Split( table.cluster, [r_threshold]|[i_numberOfClusters] )iarray of cluster numbers for each row.
make tree t matrix "upgma"
cl = Split( t.cluster, Max( t.cluster )/2 ) Split ( s_source, s_separator, regexp )sarray with the source string separated by regular expression
Split("a b \t\tc", "\s+", regexp) # returns 3-array of {"a" "b" "c"}
Split("a_asd_b_awe_c","_a.._", regexp) # returns { "a","b","c" } Split ( S_source, s_separator1, s_separator2 [regexp|exact] ) → T_tablesarray as an input. Each entry of sarray has the following syntax:
namesep2valuesep1namesep2value ...table with columns name1, name2, etc. filled with corresponding values.
Split( { "a=1;b=2", "a=3;c=5" "d=1;b=1;e=7" } ";" "=" ) Split ( X_1_with_n_molecules ) → X_n
Splits a single multimolecular entry (e.g. a compound plus water molecules) into individual
molecules. It always return at least one element.
add column t Split( Chemical( "O=O.NN" ) )Sum chemical other chemical functions
matrix option) without superposition.
Srmsd(as) → r_tzRmsd Srmsd(as,as2,[align|auto|exact|pharmacophore|type|virtual]) → r Srmsd(as_Nmol,as_Kmol,[chemical] matrix) → M_NxK_srmsds Srmsd(as_Nmol,as_Kmol,[weight] matrix) → M_NxK_superposition_errors (uses gaussian weights with TOOLS.superimposeMaxDeviation) Srmsd(as,as2,ali) → r Srmsd(ms,ms2,as_subset,chemical [,output]) → r # R_2out deviations with option Srmsd(as,as2,r_scale) → r_RelativeDisplacementErrorPerc Srmsd(X_3D as) → R Srmsd(X_3D X_3D) → M_m_n Srmsd(X_3D ) → M_n_n Srmsd(X_3D as [pharmacophore]) → R Srmsd ( ms_select1 ms_select2 chemical [output] ) Srmsd ( ms_select1 ms_select2 as_subselect1 chemical ) real root-mean-square distance between two selected chemical (hetero) molecules
according to the optimal chemical match but without 3D superposition.
With the third selection argument, the deviation will be calculated only for the as_subselect1 atoms while the equivalence pairs are established using the first two selections.
output will produce R_2out array with individual deviations for the matched pairs.
Rmsd( ms1 ms2 chemical ) and superimpose
command.
- returns Srmsd ( as_select1 as_select2 [{ align | ali } | auto|exact|pharmacophore|type|virtual] ) real value of root-mean-square deviation (returns a matrix with the matrix option ).
Similar to function Rmsd, but works without optimal superposition,
i.e. atomic coordinates are compared as they are without modification.
Number of equivalent atom pairs is saved in i_out
(see alignment options).
Virtual atoms. Be default, the first two virtual atoms ( vt1 and vt2 ) are automatically
excluded from both selections unless the virtual option is explicitly specified.
matrix option exist:
aligns amino chains by residue number and returns a [ nofMol1:nofMol2 ] matrix.
Srmsd( as1 as2 matrix ) → M_dist
performs a chemical superposition
Srmsd( as1 as2 chemical matrix ) or Srmsd( as1 as2 matrix chemical ) → M_dist
Examples:
superimpose a_1.1 a_2.1 # two similar objects, each
# containing two molecules
print Srmsd(a_1.2//ca a_2.2//ca) # compare how second molecule
# deviates if first superimposed
- returns Srmsd ( as_select [selftether] ) real root-mean-square length of absolute distance restraints
( so called, tethers ) for the tethered atoms in ICM-object.
With the selftether keyword the internal positions are used (they are set by the convert command or can be set manually).
The "tether" version is equivalent to
Sqrt(Energy("tz")/Nof(tether)) after
show energy "tz" . Similarly, the "ts" terms can be used for the selftethers.SRMSD and a full distance matrix for all pairs of molecules.i_out and the maximal deviation in
r_2out .
Srmsd ( as1 as2 matrix [chemical] ) → M_nMol1_nMol2Nof(Mol( as1 )) x Nof(Mol( as2 )) .
This function will consider two residues equivalent if they have the same residue numbers and two
atoms equivalent if they have the same names.
If no equivalences are found the srmsd value of 999. is returned.
Srmsd ( as1 as2 matrix ) → M_nMol1_nMol2_srmsds Srmsd ( as1 as2 matrix weight ) → M_nMol1_nMol2_percent_deviated_pairsmatrix chemical ) and for each pair of molecules.
The equivalence is found based on residue numbers and atom names and the returned values are root-mean-square-distances per pairs of molecules.
weight is specified, for each pairs of molecules the percent of pairs of equivalent atoms deviating beyond TOOLS.superimposeMaxDeviation is returned.
Then a difference measure with weight option is calculated as
n_matching = sum of exp ( - distance2 / threshold2 ) is calculated for each pair of atoms.
This sum is then subtracted from the total number of pairs and the measure is then divided by the number of atom pairs and multiplied by 100. ( n_different = n_pairs - n_matching ; perc_diff = 100.* n_different / n_pairs )
The threshold is defined in TOOLS.superimposeMaxDeviation (this parameter is also used in superimpose minimize command ).
Srmsd ( as_pharmTemplate as_chem [pharmacophore] ) real value of root-mean-square deviation of pharmacophore points between as_pharmTemplate and as_chem.read binary s_icmhome + "example_ph4.icb"
parrayToMol t_3D.mol[4949]
superimpose a_pharma. a_2. pharmacophore
display a_2.
Srmsd( a_pharma. a_2. pharmacophore )Rmsd superimpose create a pharmacophore object
Srmsd ( X_chemarray as_pharmTemplate pharmacophore ) rarray value of root-mean-square deviation of pharmacophore points between as_pharmTemplate and 3D chemicals from X_chemarray.
String() → s String() → s_empty String( as [name|number] | [simple [atom|residue|molecule|object]] ) → s_selection # see also: Name(<> full), l_showResCodeInSelection String( as all|dot|sln|smiles ) → s `String-date{String} ( date s_spec ) → s # see `Date String( i|l|r [n_decimals] |pref ) → s_ String( blob_ [ 'base64'|'hex' ] ) # blob to string. see `Blob `read-blob String( l_condition s_yes s_no ) # like this C expression l ? s1 : s2 String( alignment|macro|model|sequence ) → s String( s i_from i_len ) | (fr to s) → s_substr String( s key| hash ) (or 32 hash) → s_CRC32|s_MD5(see md5sum unix tool) String( s n_repeats ) → s_repeated String( slide gui ) → s_layoutString String( table "tex"|"html" [header] ) → s_printTable String( icm_word | unknown ) → s_ String( X_chemarray | table mol ) → s_sdfFileText String( array | format ) → s_columnFormat
- converts sequence into a String ( seq ) string
- converts integer into a String ( i ) string . see Tostring
- converts real into a String ( r [ i_nOfDecimals] ) string .
It also allows one to round a real number to a given number of digits after decimal point.
- repeat specified String ( s, i_NofRepeats ) string i_NofRepeats times
- adds flanking quotes and extra escape symbols to write this string in a form interpretable in shell in $string
expression.
String ( string, all )
- return URL-encoded version of the input string argument. See also Table-urland String ( string, html ) Collection to parse URL encoded strings.
- return URL-encoded query string from the input collection argument. See also Table-urland String ( collection, html ) Collection to parse URL encoded strings.
- return HTML5 canvas and rendering JavaScript
String ( X_chem, html )
- return inline html image representation.
String ( w_img, html )
- if the input s_input string is empty returns the s_default, otherwise returns the s_input string
String ( s_input, s_default )
- returns substring of length i_length. If i_length is negative returns substring from
the offset to the end.
String ( string, i_offset, i_length )
- converts numbers into a String ( { iarray | rarray | matrix } plot [ s_translateString ] ) string
or ascii characters (the "Ascii art", i.e. 12345 -> "..:*#").
The range between the minimal and maximal values is equally divided into
equal subranges for each character in the string. This function
is useful for ascii visualization of arrays and matrices. The default
translation string
is ".:*0#". Another popular choice is "0123456789".
Examples:
See also: file=s_tempDir//String(Energy("ener")) # tricky file name
show Index(String(seq),"AGST") # use Index to find seq. pattern
tenX = String("X",10) # generate "XXXXXXXXXX"
show String(Random(1.,10.,30), plot )
read matrix
show String(def," ..:*#") # redefine the projection symbolsTostring , show map.
- returns substring starting from i_from and ending at i_to.
If i_from is less than i_to the string is inverted.
Zero value is automatically replaced by the string length, -1 is the last but one element etc.
String ( i_from, i_to, string )
Examples:
String(1,3,"12345") # returns substring "123"
String(4,2,"12345") # returns substring "432"
String(1,0,"12345") # returns "12345"
String(0,1,"12345") # returns INVERTED string "54321"
String(-1,1,"12345") # returns "4321" String( date s_format )Date function.
Examples:
String( Date() "%A" ) # day of the week
String( Date() "%B" ) # monthDate
String( X ) → s_sdfFile mol/sdf file format.
This can be used to read one or multiple chemicals from a table into 3D objects
in ICM shell.
Example:
group table t Chemical("CC=O")
read mol input=String(t.mol[1]) # creates 3D objects
# to write as a file use write table mol t String( T mol ) → s_sdfFile mol/sdf format for the table or table selection.
All table fields are included into the result
add column t Chemical({"Cc1ccc(C)c(c1)c1c(C=C2C(N(CC(O)=O)C(=S)S2)=O)cn(c2ccccc2)n1", "COc1cccc(C=C2C(N(CCC(O)=O)C(=S)S2)=O)c1OCC=C"})
add column t Predict(t.mol,"MolLogP") name="MolLogP"
String( t[1] mol )
- converts the alignment into a multiline String ( ali ) string .
You can further split it into individual lines like "--NSGDG"
with the Split(String(ali_)) command.
The offset in a specific sequence and its number can be found as follows.
Examples:
read alignment s_icmhome+"sh3"
offs=Mod(Indexx(String(sh3),"--NSGDG"),Length(sh3)+1)
# extract alignment into a string, (+1 to account for '\n')
iSeq = 1 + Indexx(String(sh3),"--NSGDG")/(Length(sh3)+1)
# identify which sequence contains the pattern
- returns a Newick tree String ( ali tree ) string describing the topology of the evolutionary
tree. The format is described at
http://evolution.genetics.washington.edu/phylip/newicktree.html .
Example:
read alignment s_icmhome+"sh3"
show String(sh3 tree)
String ( s_ali ali_from { seq | i_seqNumber } )
- returns a projected string . The s_ali string contains characters
defined for each position of alignment ali_from. The function squeezes out the
characters which correspond to insertions into sequence seq_ .
This operation, in effect, projects the alignment string s_ali onto sequence seq_.
See also the
Rarray(R_,ali_,seq_) function to project rarrays.
Example (projection of the consensus string onto a sequence):
read alignment s_icmhome+"sh3" # 3 seq.
cc = Consensus(sh3)
show String(Spec)//String(cc,sh3,Spec)
- projects the input
String ( s_seq { seq | i_seqNumber } ali_to s_gapDefChar ) string from seq_ to ali_to (the previous function does it in the opposite direction). The R_seq string contains characters
defined for each position of the sequence seq_. The function fills the gap positions in the
output with the r_gapDefChar character.
Combination of this and the previous functions allow you to project any string s1 from
one sequence to another by projecting the s1 of seq1 first to the alignment and than back to
seq2 (e.g. String( String(s1,seq1,a,"X") , a, seq2) ).
See also the
Rarray( R_,seq_,ali_,r_gapDefault ) function to project real arrays.
Example (transfer of the secondary structure from one sequence to another):
read alignment s_icmhome+"sh3" # 3 seq.
ssFyn = Sstructure(Fyn)
set sstructure Spec String(String(ssFyn,Fyn,sh3,"_"),sh3,Spec)
show Spec
String( l_condition s_choice1 s_choice2 )a=3
String( a>1 , "big a", "small a" ) String( { os | ms | rs | as } [ name | number ] [ i_number ] ) String( { os_1 | ms_onOneObj } simple ) a_a.1:4|a_b.2,14 )
You can also divide this selection info a string array with the Split function.
Option i_number allows one to print only i-th element of the selection.
It is convenient in scripts. For atom selections it will also show
full information about each atom, rather than only the ranges of atom numbers.
This string form is convenient used for several purposes:
Select function)
With option name , the one-letter residue code will be shown in addition to the number, regardless of the l_showResCodeInSelection system logical ( e.g. a_/^F23 instead of a_/23 ). Conversely option number shows only the number.
By default (without the name option) the code is shown depending on the l_showResCodeInSelection flag.
simple : a special function that returns the selection in terms of the name of a single object (make sure that only one object is selected) and
name | number : allows one to show or hide, respectively, the one-letter code of a residue (e.g. '^W123').
the names of the molecules (e.g. a_1abc.a ). See also the Name function to get object of molecule names (e.g. Name( a_1. ) ↑ "1abc" )
An example in which we generate text selection of the Crn leucine neighbors :
Another example with a loop over atom selection of carbon atoms:
nice "1crn"
l_showResCodeInSelection = no
nei = String( Res(Sphere( a_/leu a_/!leu , 4.)) )
show nei
a_1crn.a/14:17,19:20
display xstick $neiread pdb "2ins"
for i=1, Nof( a_//c* )
print String( a_//c* i )
endfor
See also: l_showResCodeInSelection
String( slide gui ) → s_layoutStringsl = Slide(gui)
undisplay window="all"
# take a look
display window=String( sl gui )Slide, display window.
String ( as { dot | all | smiles | sln } )
- returns string with the following chemical information:
option description example
all chemical formula e.g. C2H6O
dot chemical formula with dot-separated molecules e.g. C2H6O.C3H8
smiles smiles string e.g. [CH3][CH2][OH]
sln sln notation e.g. CH3CH2OH
Molecules with a certain chemical formula (calculated without hydrogens)
can be selected by the a_formula1,formula2.. selection .
See also: Smiles , smiles , selection by molecule.
Example:
build string "se ala" # alanine
show String(a_//!h* all ) # returns no hydrogen chemical formula: C3NO
show String(a_//* all ) # returns chemical formula: C3H5NO
show String(a_//* sln ) # returns SLN notation: NHCH(CH3)C=O
show String(a_//* smiles) # returns SMILES string: [NH][CH]([CH3])C=O
- returns Sstructure ( rs ) string
of secondary structure characters ("H","E","_", etc.) extracted from specified residues `rs_ .
- returns the compressed Sstructure ( { rs | s_seqStructure } compress ) string
of secondary structure characters, one character per secondary structure
segment, e.g. HHE means helix, helix, strand.
Use the
Replace function to change B to '_' and
G helices to H helices, or simply all non H,S residues to coil
(e.g. Sstructure(Replace(ss,"[!EH]","_"),compress) )
Example:
show Sstructure("HHHHHHH_____EEEEE",compress) # returns string "HE"
#
read object "crn"
show Sstructure( a_/A , compress) # returns string "EHHEB"
- returns Sstructure ( { seq | s_sequenceString } ) string
of secondary structure characters ("H","E","_").
If this string has already been assigned to the sequence seq_ with the
set sstructure command or the
make sequence ms_ command, the function will return
the existing secondary structure string. To get rid of it, use the
delete sstructure command.
Alternatively, if the secondary structure is not already defined,
the Sstructure function will predict the secondary structure of the seq_
sequence with the Frishman and Argos method.
If the specified sequence is not a part of any
alignment of sequence group only a single sequence
prediction will be effected (vide infra). Otherwise, a group or an alignment
will be identified and a true multiple sequence prediction algorithm is applied. The
multiple sequence prediction by this method reaches the record of 75% prediction accuracy
on average for a standard selection of 560 protein chains under rigorous jack-knife
conditions. The larger the sequence set the better the prediction. Prediction accuracy
for a single sequence is about 68%. To collect a set perform the fasta search (
Pearson and Lipman, 1988
) with ktup=1 and generate a file with all the sequences in a fasta format.
Method used for derivation of single sequence propensities.
Seven secondary-structure related propensities are combined to produce the final
prediction string. Three are based on long-range interactions involving potential
hydrogen bonded residues in anti-parallel and parallel beta sheet and alpha-helices.
Other three propensities for helix, strand and coil, respectively, are predicted by the
"nearest neighbor" approach (
Zhang et al., 1992
), in which short fragments with known secondary structure stored in the database
(icmdssp.dat) and sufficient similarity to the target sequence contribute to the
prediction. Finally, a statistically based turn propensity (also available separately
via the
Turn( sequence)
function), is employed over the 4-residue window as described by
Hutchinson and Thornton (1994).
The function also returns four real arrays in the
M_out
matrix
[4, seqLength]. There arrays are:
Note that these propensities are not directly related to the prediction.
Usually the reliability level of 0.8 guaranties prediction accuracy of about 90%.
Do not be surprised if the propensities are all zero for a fragment. It may just
mean that the statistics is too scarce for a reliable estimate.
- returns Sstructure ( seqarray ) sarray of secondary structure strings stored in a sequence parray
show Index(Sstructure(a_1crn.,"HHHHHH")) # first occurrence of
# helix in crambin
read sequence "sh3" # load 3 sequences (the full name is s_icmhome+"sh3")
show Sstructure(Spec) # secondary structure prediction for one of them
show Sstructure("AAAAAAAAAAAAA") # sec. structure prediction for polyAla
read sequence "fasta_results.seq"
group sequences a unique 0.05 # remove redundant sequences
show Sstructure(my_seq_name) # the actual prediction, be patient
plot number M_out display # plot 3 propensities and reliability
- returns the Sum ( iarray ) integer sum of iarray elements.
- returns the Sum( { rarray | map }) real sum of elements.
- returns the Sum ( matrix ) rarray of sums in all the columns.
- returns Sum ( sarray [ s_separator ] ) string of concatenated components of a sarray separated by
the specified s_separator or blank spaces by default.
See also the opposite function: Split .
Examples:
show Sum({4 1 3}) # 8
show Sum(Mass(a_1//*)) # mass of the first molecule
show Sum({"bla" "blu" "bli"}) # "bla blu bli" string
show Sum({"bla" "blu" "bli"},"\t") # separate words by TAB
show Sum({"bla" "blu" "bli"},"\n") # create a multiple line string
>>Sum-multisep
h4-- Concatenate multiple columns
Sum( T_table { S_cols } s_sep1 s_sep2 ) → S_resultsarray where each element formed as follows:
colname1sep2value1sep1colname2sep2value2 ...add column t {"" "a" "" "" "b"} {"c" "" "j" "r" ""} {"a" "u" "" "" "b"}
Sum( t ":", "=" )Split multisep
Sum ( chemarray [r_spacing=0.] )read pdb "1zkn"
read mol input=String(Sum( Chemical( a_ibm,ibm2 exact )) ) split name="twoMolsInOne"Split chemical other chemical functions
Sum ( imageArray_a imageArray_b [r_bweight(0.5)] )Image
- returns the Symgroup ( { s_groupName | os_object | i_groupNumber | m_map } number ) integer number of one of 230 named space groups defined in ICM.
- returns the Symgroup ( { i_groupNumber | os_object | m_map } ) string name of one of 230 space groups defined in ICM.
The number of transformations for all two versions is returned in i_out .
Transform :
- returns the Transform ( i_groupNumber ) rarray of transformation matrices (12 numbers each) describing symmetry operations
of a given space group.
Examples:
iGroup = Symgroup("P212121" number ) # find the group number=19
print "N_assymetric_units in the cell =", i_out
show Transform(iGroup)) # shows 4 12-membered transformations.
Table(ali I_aliPos residue|label) → T_posNumbers `alignment-as-table{Table} (ali [number]) → T_seqColumns Table( map ["min"|"max"] ) → T_x_y_z_value `Table-matrix{Table} (matrix_nxm [S_colnames_m]) → T_with_m_columns `Table-pairs{Table} ( matrix_nxm [ S_rowtags_n S_coltags_m ] index ) → T_nm_ij_pairs Table( model term|merit) → T_statReport Table( T S_Tcolnames) → T_columnSubseletion Table( pairdist distance ) → T_atomsPairs Table( parray [s_colName]) → T_with_parray_column Table( collection ) → T_converted_from_the_collection Table( residue ) → T_icm_res_names_codes Table(seq) → T_resContent (name,n,freq) Table(seq site) → T_siteInfo (key,fr,to,list,desc) Table( stack [vs] ) → T_confTerms Table( s_svgTextNodesEdges plot ) → T_nodes_edges `Table-url{Table} (URL_encoded_string_a=b&c=d&e=ff [crypt] ) → T_name_valueicm.res file loaded by the read library command with the following columns:
Example:
.char .name .type .desct = Table(residue)
tt = t.type=="Amino"
show tt # currently loaded amino acids
if Index(tt.name,"tyr") != 0 print "legal residue name"
Table ( s_URL_encoded_String [ crypt ] )
- returns the table of "name" and "value" pairs organized in two
string arrays.
The URL-encoding is a format in which the HTML browser sends the HTML-form input
to the server either through standard input or an environmental variable.
The URL-encoded string consists of a number of the "name=value&name=value..."
pairs separated by ampersand ( & ). Additionally, all the spaces are
replaced by plus signs and special characters are encoded as hexadecimals with
the following format %NN. The Table function decodes the string and creates
two string arrays united in a table.
Option crypt allows one to interpret doubly encoded strings
(e.g. ' ' is translated to '+' which then converted into a
hexadecimal form). Frequently the problem can be eliminated by specifying
the correct port. Example: you need to set a="b c" and d="<%>".
Normal server will convert it to a=b+c&d=%3C%25%3E.
Double encoding leads to a=b%2bc&d=%253C%2525%253E.
To parse the last string, use the crypt option.
To see all the hidden symbols (special attention to '\r'),
set l_showSpecialChar =yes.
Examples:
See also:
Getenv( ).
read string # read from stdin in to the ICM s_out string
a=Table(s_out) # create table a with arrays a.name and a.value
show a # show the table
for i=1,Nof(a) # just a loop accessing the array elements
print a.name[i] a.value[i]
endfor
Table ( alignment [ number ] ) table of relative amino acid positions for each of the sequence
in the alignment. Gaps are marked by zero.
Note that here columns correspond to different sequences while rows correspond to alignment positions.
In the next function this order is reversed.
The first column of the table, .cons , contains sarray of consensus characters.
All the other arrays are named according to the sequence names by default, or
by the sequential number of a sequence in the alignment, if option number is specified.
The table may be used to project numbers from one sequence to another.
See also the Rarray( R_, ali_, seq_ ) function.
#>T pos
#>-cons----seq1-----seq2-------
" " 0 1
" " 0 2
C 1 3
" " 2 0
~ 3 4
C 4 5
" " 0 6
# for the following alignment:
# Consensus C ~C
seq1 --CYQC-
seq2 LQC-NCP
To calculate an array of mean scores for each column of a multiple sequence alignments use the Rarray( ali [ exact ] ) function.
This array can be appended to the table.
Example:
See also, the next function
read alignment "sh3"
t = Table(sh3 number) # arrays t.1 t.2 t.3
t = Table(sh3) # arrays t.cons t.Fyn t.Spec t.Eps8
#
cc = t.cons ~ "[A-Z]" # all the conserved positions
show cc # show aa numbers at all conserved positions
show t.Fyn>=10 & t.Fyn<=20 # numbers of other sequences in this range
Table( alignment, I_alignmentPositions , residue | label )
returns a table of corresponding residue numbers for the selected positions
I_alignmentPositions . With option residue only the numbers are returned,
while under the label option, the residue labels (e.g. Y25 ) are returned.
p11, p12 .. )
Table( aaa {1 11 13 16} label ) # aaa contains three sequences
seq p1 p11 p13 p16 comment 1fyn_a L10,90 D12,92 A15,95 this sequence is linked to molecule a
Spec D1 L11 D13 E16 Spec sequence positions starting from 1
Eps8 K1 K11 D13 A16 Eps8 sequence positions starting from 1
Table ( matrix [ S_colnames ] ) → Ttable with matrix columns named 'A', 'B', .. or according to the second argument.
Example:
t= Table(Matrix(3),Sarray(3,"A")+Count(3))
show t
>T t
#>-A1---A2---A3----
1. 0. 0.
0. 1. 0.
0. 0. 1. Table(matrix_nxm [S_rowtags_n S_coltags_m] index ) → T_nm_ij_pairsI,J,C or A,B,I,J,C containing a two indexes and (if provided) two names of elements and their Mij value.
It will return all values.
It the
build string "ala"
m=Distance(Xyz(a_//c*) Xyz(a_//c*) ) # carbon distance matrix
ats = Sarray( a_//c* )
t = Table(m ats ats index)
add column t t.I-t.J name={"D"}
delete t.D<=0 # get rid of the diagonal and lower triangle
show t
# another example:
m = Random(2,4,1.,3.) # matrix 2x4
r= {'a','b'} # row tags
c= {'u','x','y','z'} # col tags
Table( m r c index)
Table( stack [ vs ] )
- return table of parameters for each conformation in a stack .
If a variable selection argument is provided, the values
of the specified variables are returned as well.
% icm
build string "ala his trp"
montecarlo
show stack
iconf> 1 2 3 4 5 6 7
ener> -15.1 -14.6 -14.6 -14.2 -13.9 -11.4 -1.7
rmsd> 0.3 39.2 48.0 44.1 27.4 56.6 39.3
naft> 1 0 0 1 1 1 0
nvis> 4 1 1 4 4 4 1
t= Table(stack)
show t
#>T t
#>-i--ener--------rmsd--------naft--------nvis-------
1 -15.126552 0.295555 1 4
2 -14.639667 39.197378 0 1
3 -14.572973 47.996203 0 1
4 -14.220515 44.058755 1 4
5 -13.879041 27.435388 1 4
6 -11.438268 56.636246 1 4
7 -1.654792 39.265912 0 1
t1= Table(stack v_//phi,psi) # show also five phi-psi angles
#>T
#>-ener----rmsd--naft-nvis------v1------v2------v3------v4-------v5-----
1 -15.12 0.29 1 4 -79.10 155.59 -75.30 146.99 -141.13
2 -14.63 39.19 0 1 -157.22 163.56 -78.25 139.51 -137.30
3 -14.57 47.99 0 1 -157.26 166.87 -85.08 92.55 -84.74
4 -14.22 44.05 1 4 -67.65 80.43 -76.67 103.05 -81.85
5 -13.87 27.43 1 4 -82.72 155.86 -85.02 93.11 -81.46
6 -11.43 56.63 1 4 -78.28 152.80 -154.79 66.26 -77.61
7 -1.65 39.26 0 1 -78.17 169.41 -133.89 96.39 -96.03
See also: Iarray stack function
Table ( s_graphviz_svg plot ) → T_nodes_edges_for_resortingneato tool from the graphviz dot package and parses it into
rows for resorting to solve the problem of lines overlapping the nodes.
The table contains the following columns:
i : original order
tx : svg text corresponding to header/footer or a node/edge row
ty : type of the row: one of four: "begin","node","edge","tail"
width : a number for sorting. Attempts to find tags: stroke-width and put thick lines on top.
read string "/tmp/sgraph.svg" name="svg" # original svg with overlaping edges.
tsvg = Table(svg, plot)
sort tsvg.width
write Sum(tsvg.tx) "/tmp/sgraph_sorted.svg"
Table( plsModelName [ term | merit ] ) name mean rmsd, -w (weight) and -wRel
columns.
The header of the table contains the free term ( constant b ).
The linear model can be represented as
Ypred = b + w1*X1+w2*X2+...The wRel column returns the following value:( Abs(wk) * Rmsd(Xk) ) / Sum_k( Abs(wk) * Rmsd(Xk) )A = Random(1. 10. 20)
group table T A A*2. "B" Random(1. 10. 20) "C" Random(1. 10. 20) "D"
write binary Apred
delete Apred
#
read binary "Apred"
Table( Apred term )
#>r .b
0.012402
#>r .self_R2
0.999998
#>r .test_R2
0.999885
#>r .self_rmse
0.002908
#>r .test_rmse
0.030066
#>T
#>-name--------mean--------rmsd--------w-----------wRel-------
B 11.04767 4.620291 0.499992 99.726648
C 5.749607 2.681675 -0.001182 0.13679
D 4.686537 2.686346 -0.001178 0.136562 Table( s_buildInModel|F_model X_chemarray [inverse] )
tt = Table( "MolLogP", Chemical( "CCO" ) )
tt.ch_1 != 0
Table( "MolLogP", Chemical( "OOO" ) inverse )
tt_stat = Table( myModel, tt.mol inverse ) Table( hbondpairs|atompair_distances|angles|torsions distance ) → T_atomsPairsdistance object and returns a table with the following columns
Angles and torsions will also have atom1 # selection , e.g. a_a.b/^T3/cn
atom2 # second atom
dist # distance in Angstroms
color # color if present
label # label of this distanceatom3 and atom4 columns.
read pdb "1crn"
convertObject a_ yes yes no no
make hbond name="hbonds_crn"
show Nof( hbonds_crn ) # counts distances
t = Table( hbonds_crn distance )
sort t.dist
show t[1]make distance , make hbond , Nof-distance{Nof(d,distance)}
Table ( chemical charge ) table with concentrations of various prononation state vs pH
Table( chemical r_pH r_concCutoff charge )table with protonation states at given ph4 and percentage cutoff
- returns the Tan ( { r_Angle | i_Angle } ) real value of the tangent of its real or integer argument.
- returns Tan ( rarray ) rarray of the tangents of each component of the array.
Examples:
show Tan(45.) # 1.
show Tan(45) # the same
show Tan({-30., 0. 60.}) # returns {-0.57735, 0., 1.732051}
- returns the Tanh ({ r_Angle | i_Angle } ) real value of the hyperbolic tangent of its real or integer argument.
- returns Tanh ( rarray ) rarray of the hyperbolic tangents of each component of the array.
Examples:
show Tanh(1) # returns 0.761594
show Tanh({-2., 0., 2.}) # returns -0.964028, 0., 0.964028
Tensor ( M)
- returns the square matrix of second moments of K points in N -dimensional space,
Mki (k=1,K,i=1,N) .
The matrix NxN is calculated as
< Xi >< Xj > - < Xi Xj >
, where < .. > is averaging over a column k=1,K, and i,j=1,N.
If xyz is a coordinate matrix Nx3, the Tensor function is identical to
Transpose( xyz ) * xyz / Nof(xyz)
Eigen or Disgeo functions.
This trick used in the dockScan script ( _dockScan.
Example:
This commands are assembled in the build string "AAA" # a long molecules
xyz = Xyz( a_//c* ) # a coordinate matrix of carbons
# you can also do it with grobs: xyz = Xyz( g_myGrob )
a=Tensor(xyz) # compute 3 by 3 matrix of the second moments
b=Eigen(a) # returns 3 axis vectors
ax1= b[?,1] # this is the longest half axis
ax2= b[?,2] # this is the second half axis
ax3= b[?,3] # this is the shortest half axis
len1 = Length(ax1) # long axis length
len2 = Length(ax2) # mid axis length
len3 = Length(ax3) # short axis length
r = Matrix(3,3)
# to make the rotation matrix from b normalize the axes
r[?,1] = ax1 / Length( ax1 )
r[?,2] = ax2 / Length( ax2 )
r[?,3] = Vector( r[?,1], r[?,2] )
rotate a_ Transpose(r) # rotates the principal axes to x,y,z
# x the longestcalcEllipsoid M_xyz macro which returns
ellipseRotMatrix , and three vectors: ellipseAxis1 , ellipseAxis2 and ellipseAxis3
See also:
Rot, rotate, transform
<>
Example to orient the principal axes of the molecule along X,Y and Z
(the longest axis along X, etc.).
build string "se ala ala ala ala" # let is define the ellipsoid
display virtual
a = Tensor(Xyz(a_//!h*)) # Xyz returns matrix K by 3
b=Eigen(a) # 3x3 matrix of 3 eigenvectors
b[?,1] = b[?,1] / Length( b[?,1] ) # normalize V1 in place
b[?,2] = b[?,2] / Length( b[?,2] ) # normalize V2
b[?,3] = Vector( b[?,1], b[?,2] ) # V3 is a vector product V1 x V2
rotate a_ Transpose( b ) # b is the rotation matrix now
# Transpose(b) is the inverse rotation
set view # set default X Y Z view
Temperature ( { s_DNA_sequence | seq_DNA_sequence } [ r_DNA_concentration_nM [ r_Salt concentration_mM ] ] )
- returns the real melting temperature of a DNA duplex at given
concentration of oligonucleotides and salt. The temperature is
calculated with the Rychlik, Spencer and Roads formula
(Nucleic Acids Research, v. 18, pp. 6409-6412)
based upon the
dunucleotide parameters provided in Breslauer, Frank, Bloecker,
and Markey, Proc. Natl. Acad. Sci. USA, v. 83, pp. 3746-3750.
The following formula is used:
Tm=DH/(DS + R ln(C/4)) -273.15 + 16.6 log[K+]
where DH and DS are the enthalpy and entropy for helix formation,
respectively, R is the molar gas constant and C is the total molar
concentration of the annealing oligonucleotides when oligonucleotides are not self-complementary. The default concentrations are C=0.25 nM and
[K+]= 50 mM. This formula can be used to select
PCR primers and to select probes for chip design.
Usually in primer design the
temperatures do not differ from 60. by more than several degrees.
- returns the Time ( string ) string of time (e.g. 00:12:45 ) spent in ICM.
- returns the Time ( ) real time in seconds spent in ICM.
Examples:
if (Time( ) > 3660.) print "Tired after " Time(string) " of work?"
- converts to Tointeger ( string|real|integer|logical )integer
- converts each element to integer, returns Tointeger ( sarray|rarray|iarray|array )iarray.
- maps real numbers from the R_source to integers.
The R_splitPoints array of a size n should contain numbers in increasing order.
Those n points will be used as split points for n+1 intervals.
I_values of size n+1 specifies numbers to be assigned to values in each of those intervals.
Tointeger ( R_source R_splitPoints I_values ) classification problems .
Tointeger( {-1., -2., 3. 4. 5. 6.},{0.},{-1,1} )
{-1, -1, 1, 1, 1, 1}Tointeger({1. 2. 3. 4. 5. 6.},{2.5,4.5},{2,4,6}) Tointeger ( S_source S_labels I_values ) Tointeger ( I_source I_labels I_values )Tointeger( {"dit" "dah" "dah" "dah" "dit" "dah"} {"dit" "dah"} {0 1} )
0 1 1 1 0 1
Tointeger( {"dit" "dah" "dah" "XXX" "dit" "dah" "YYY" "dah"} {"dit" "dah"} {0 1 100} )
0 1 1 100 0 1 100 1
Tointeger( {1 5 1 5 6 7 6 1} {1 5} {2 3 0} )
2 3 2 3 0 0 0 2Toreal( ), Tostring( )
- returns the Tolower ( string ) string converted to the
lowercase. The original string is not changed
- returns the Tolower ( sarray ) sarray converted to the
lowercase. The original sarray is not changed.
Examples:
See also:
show Tolower("HUMILIATION")
read sarray "text.tx" #create sarray 'text' (file extension is ignored)
text1 = Tolower(text)Toupper( ).
- converts to Toreal ( string|real|integer )real
- converts each element to a real, returns a real array. see Toreal ( sarray|rarray|iarray )Rarray( ).
- converts each key to a respective real value. If values contains n+1 elements,
the last value is the deault value (used to convert all keys not in keys). Toreal ( S S_n_keys R_n1_values ) # R_values has n or n+1 elementsToreal({"c","a","c","c"},{"c","a"},{1,2}) # two classes
{1, 2, 1, 1}
Toreal({"c","a","q","c","k"},{"c","a"},{1.8,2.3,0.5}) #with default value 0.5
{1.8, 2.3, 0.5, 1.8, 0.5}rarray constant describes special values in real arrays that may appear in real columns of tables upon reading the Excel/csv files
or property fields of the mol (or sdf) .
Example:
Create file 't.csv' that looks like this:
To compare an array with special values with a specific special value use this:
1.1
ND
3.3
INF
>3.
<2.read csv "t.csv"
t.A == Toreal({"ND"})
t.A != Toreal({"ND","INF"})
t.A == Toreal({">3."})Tointeger( ), Tostring( )
- returns the Torsion ( as ) real torsion angle defined by the specified atom as_
and the three previous atoms in the ICM-tree.
For example, Torsion(a_/5/c) is defined by
{ a_/5/c , a_/5/ca , a_/5/n , a_/4/c } atoms.
You may type:
print Torsion( and then click the atom of interest, or use GUI to calculate the angle.
- returns the Torsion ( as_atom1, as_atom2, as_atom3, as_atom4 ) real torsion angle defined by four specified atoms.
Examples:
d=Torsion( a_/4/c ) # d equals C-Ca-N-C angle
print Torsion(a_/4/ca a_/5/ca a_/6/ca a_/7/ca) # virtual Ca-Ca-Ca-Ca
# torsion angle
- converts to Tostring ( string|real|integer )string
- converts each element to a string, returns Tostring ( sarray|rarray|iarray )sarray.
- returns Tostring ( seqarray )sarray with sequences extracted from sequence parray elements.
Toreal , Tointeger , Sequence
- returns the Toupper ( string ) string converted to the uppercase. The original string is not changed
- returns the Toupper ( sarray ) sarray converted to the uppercase. The original sarray is not changed.
Toupper ( string|sarray 1 )
Examples:
See also:
show Toupper("promotion")
show Toupper("joseph louis gay lussac",1)
Joseph Louis Gay-Lussac
read sarray "text.tx"
text1 = Toupper(text)Tolower( ).
- returns Tr123 ( sequence ) string like "ala glu pro".
Examples:
See also:
show Tr123(seq1)Tr321( ).
IcmSequence( ).
- returns Tr321 ( s ) sequence
from a string like this: "ala glu pro". This function is
complementary to function
Tr123( ). Unrecognized triplets will
be translated into 'X'.
Examples:
show Tr123("ala his hyp trp") # returns AHXT
- returns the Trace ( matrix ) real trace (sum of
diagonal elements) of a square matrix.
Examples:
show Trace(Matrix(3)) # Trace of the unity matrix [3,3] is 3.
- extracts the R_3
vector of translation from the
transformation vector.
Trans ( R_12transformationVector )
Trans ( seq_DnaOrRnaSequence )
- returns the translated DNA or RNA sequence ('-' for a
Stop codon, 'X' for an ambiguous codon) using the
standard genetic code.
See also:
Sequence( seq_ reverse ) for the reverse complement DNA/RNA sequence.
Example (6 reading frames):
w=Sequence("CGGATGCGGTGTAAATGATGCTGTGGCTCTTAAAAAAGCAGATATTGGAG")
show Trans(w), Trans(w[2:999]),Trans(w[3:999])
c=Sequence(w,reverse)
show Trans(c), Trans(c[2:999]),Trans(c[3:999])
Trans ( seq_DnaOrRnaSequence { all | frame } [ i_minLen] [ s_startCodons] )
return a table
of identified open reading frames in DNA sequence not shorter than i_minLen .
The function was designed for very large finished sequences from the genome
projects. Currently the Standard Genetic code is used.
Option s_startCodons allows one to provide a comma-separated list of starting codons;
if omitted, the default is "ATG" , another example would be "ATG,TTG"
(for S.aureus).
Option frame indicates that both start and stop codons need to be found.
If they is not found or the fragment is too short, the table will be empty.
Option all allows one to translate ALL POTENTIAL peptides by assuming
that the start and/or stop codons may be beyond the sequence fragment. In this
case, initially all 6 frames are produces. Later, some of them can be filtered out
by the i_minLen threshold. The unfinished end codons will be marked by 'X'.
The table has the following structure:
For example, if the fragment is in the complementary strand it may have
the following parameters:
In this case translation follows the reverse strand (frame=-1),
starts in position 57 of the original direct sequence and proceeds
to position 22.
#>-frame-------left--------right-------dir---------len---------seq--------
-1 22 57 -1 12 XCVXVAAESVAS
Example:
dna=Sequence("TTAAGGGTAA TATAAAATAT AAAGTTCGAA CAATACCTCA CTAGTATCAC AACGCATATA")
T=Trans(dna frame 10)
sort T.left
show T
Transform( s_group|iGroup|os_1|map ) → R_12N_all_fract_transformations Transform( s_group|iGroup|os_1|map iTrans ) → R_12_fract_transformation_i Transform( s_group iTrans R_6cell ) → R_12_abs_transformation_i Transform( obj "bio" i_biomol ) → R_12N_abs_BIOMT_transformations Transform( s_symbolic_transformation ) → R_12 # not ready Transform( R_6 ) → R_12 Transform( M_4x4 | M_3x3 ) → R_12_transformation Transform( R_12 inverse ) → R_12_inverse_transformationa_ for
the current object). The crystal symmetry and the biological symmetry can be imposed
with the set symmetry command.
- converts the argument matrix[n,m] into the transposed Transpose ( matrix ) matrix [m,n]
- converts real vector [n] into a one-column
Transpose ( rarray ) matrix [n,1]
Examples:
Transpose(a) # least squares fit
Transpose({1. 2. 3.}) # [3,1] matrix
- converts the argument table[nrows,ncols] into the transposed Transpose( table [i_nameColumn] )table [ncols,nrows]
iarray, rarray or sarray.
read sequence "seqs"
group sequence "a"
align a
t = Table( a Count(Length(a)) label )
t2 = Transpose( t 1 ) # transpose and use first column values as result column names
(t2.Azur_Alcfa == "-").Azur_Alcde # get residue labels in Azur_Alcde which corresponds gaps in Azur_Alcfa
Trim ( R [ r_percentile [ i_mode ]] )
- returns rarray of softly trimmed values. The obvious outliers are softly moved closer to the expected distribution.
This is a clever auto-trim which identifies outliers defined
as values beyond the limits [a,b] projected from range of
the r_percentile values adjusted to 100% with 10% of additional margin.
The values within the limits are not changed but the outliers are brought
closer to the majority bounds. If i_mode is 0, the outliers are assigned
to the boundary values. If i_mode is 1, the values outside the range
are scaled down according to this formulae: d_new = b + log(1.+(d-b)/(b-a))
for high values and similarly for low values.
Return values:
By default r_percentile is 0.9 and i_mode is 0 .
Example:
r_out and r_2out values.
i_out .
Trim({0. 1. 4. 6.}) # keeps values unchanged
Trim({0. 1. 4. 6. 55.},0.9,1) # returns {0. 1. 4. 6. 11.3}
Trim({-33. 0. 1. 4. 6. 55.},0.9,1) # returns {-3.5 0. 1. 4. 6. 11.3}
- a linear transformation to the [0., 1.] range, useful for generating a rainbow color index.
The Trim ( R rainbow|fix ) fix option transforms to a fixed range of [-1.,1.], useful for machine learning.
- returns Trim ( I_iarray i_lower i_upper ) iarray clamped into the specified range. Values smaller than
i_lower are replaced with i_lower, and values greater than i_upper
are replaced with i_upper.
- returns Trim ( R_rarray r_lower r_upper ) rarray clamped into the specified range.
- returns Trim ( i i_lower i_upper ) integer clamped into the specified range (e.g. Trim(6,1,3) returns 3).
- returns Trim ( r r_lower r_upper ) real clamped into the specified range.
- returns Trim ( M_matrix r_lower r_upper ) matrix clamped into the specified range.
- returns Trim ( m_gridMap r_lower r_upper ) map clamped into the specified range. It means that all values above r_upper are set to r_upper, and all values below r_lower are set to r_lower.
- returns Trim ( string|S [ all | print ] ) string (or sarray) with removed trailing blanks and carriage returns. If option all is specified,
both leading and trailing blank characters will be removed.
With print option the non-printable characters will be removed or replaced by similar printable characters.
- trims to the maximal number of characters, it may appends specified trailing string if truncated.
Trim ( string maxNofCharacters [s_appendWhenTruncated] ) Trim("123456",3) # returns "123"
Trim("123456",33) # returns "123456"
Trim("123456",3,"..") # returns "123.."
Trim ( string s_allowed_characters ) string with in which only the allowed characters are retained.
All other characters are removed.
Example:
Trim("as123d","abcds")
asd Trim ( string S_regularExpressionsToDelete ) string in which all listed regular expressions are deleted.
Trim ( S_sarray [ all ] ) sarray of strings with removed trailing blanks.
With option all it removes white space characters from both ends.
Trim( X [s_smarts ('[$([*;D1]~[*;R0])]') [i_maxSteps(999) i_minAtomsLeft (0)] ] ) → X_trimmed
Examples:
D1 will stop when there is a single carbon left, thus it will never go to zero
add column t Chemical({"C1C(CCNC)CNC1CCCC","",""} )
t.mol[2] = Trim( t.mol[1],"[*;D1]" )
t.mol[3] = Trim( t.mol[1] ) # the default will leave one attached atom
Trim ( seq S_tagRegexps ) → seq_truncatedS_proteinTags that contains popular expression tags:
Feel free to modify it or provide your own list or fragments to be deleted.
Example:
^.{0,11}HHHHHH
^.{0,5}HHHHH
^.{0,5}DYKDDDDK
DYKDDDDK.{0,3}$
HHHHHH.{0,6}$
YPYDVPDY.{0:3}$
AWRHPQFGG$read pdb sequence "1pme" # contains his-tag
cleanseq = Trim(1pme_a S_proteinTags ) # built in shell array
Align(1pme_a cleanseq)
1pme_a MSSSHHHHHHSSGLVPRGSHMAAAAAAGAG
cleanseq ----------SSGLVPRGSHMAAAAAAGAG
- returns Turn ( { seq | rs } ) rarray containing beta-turn prediction index.
The index is derived from propensities for i,i+1,i+2,i+3 positions for each
amino-acid. Pi = pi+pi1+pi2+pi3, then high Pi values are assigned to the next
three residues. The propensities are taken from
Hutchinson and Thornton (1994).
Examples:
See also the s = Sequence("SITCPYPDGVCVTQEAAVIVGSQTRKVKNNLCL")
plot comment=String(s) number Turn(s) display # plot Turn predictionpredictSeq
macro.
- returns a Type ( icm_object_or_keyword ) string
containing the object type
(e.g. Type(4.32) and Type(tzWeight) return string "real" ).
The function returns one of the following types:
"integer", "real", "string", "logical", "iarray", "rarray", "sarray","table", "aselection","vselection","sequence", "alignment", "profile", "matrix", "map", "grob", "command", "macro", "unknown".
- returns the Type (parray , 1 ) parray element type, like "mol" or "model".
- returns a Type ( as , 1 ) string containing the level of the selection ("atom","residue","molecule","object").
Type ( os_object , 2 ) string (or an sarray with keyword object) containing the os_object (or current
by default) molecular object type. Defined types follow the EXPDTA (experimental data) card
of PDB file with some exceptions, see below:
The non-ICM types can be converted to "ICM" ready for energy calculations. Those objects are either built in ICM or converted to the ICM-type.
"X-Ray" determined by X-ray diffraction
"NMR" determined by NMR
"Model" theoretical model (watch out!)
"Electron" determined by electron diffraction
"Fiber" determined by fiber diffraction
"Fluorescence" determined by fluorescence transfer
"Neutron" determined by neutron diffraction
"Ca-trace" upon reading a pdb, ICM determines if an object is just a Ca-trace.
"Simplified" special object type for protein folding games.
"ICM" with the convert command or convertObject macro.
set type object command, e.g.
set type a_ "NMR"
- returns the Type ( { ms | rs }, 2 ) string type of the specified molecule or residue.
Legal types are "Amino", "Hetatm", "Nucl", "Sugar", "Lipid", "empty".
Residues of the "Amino" type can be selected with the 'A' character
(e.g. a_/A). See also a one-letter code for the type which is used
in selections, ( e.g. a_A,H ).
set type a_2 "H" to switch to a heteroatom type.
Examples:
See also:
if (Type(a_1.1)!="Amino") goto skip: # deal only with proteins
if (Type( ) == "NMR") print "Oh, yes!"
or
Type( ms molecule|all )
for an array of one-letter molecule types or array of residue types
Type( rs residue|all )
- returns an Type ( as { atom | mmff } ) iarray containing the ICM or MMFF atom types. Example:
build string "his ala"
show Type(a_//!vt* atom ) # icm types for non-virtual atoms
- returns an Type ( as_1 as_2 ) integer containing the covalent bond type between the selected atoms.
Type( os|ms|rs all|object|molecule|residue ) → S_types all it will determine the level of selection from the selection.
Otherwise the level will be determined by the keyword. For molecules one-letter type code will be returned, e.g. "H" or "A" etc.
- Type( Type( seq , 1|2 ) string type of the sequence. Two types are recognized with option 1:
"protein" and "nucleotide" .
An example in which we rename and delete all DNA sequences from the session:
- Type( read pdb sequence "1dnk"
Type( 1dnk_b, 1 )
nucleotide
for i=1,Nof(sequence)
if Type(sequence[i],1) == "nucleotide" rename sequence[i] "dna"+i
endfor
delete sequence "dna*"a = Sequence("AATTGGC", nucleotide)
Type(a 2)
"dna"
b = Sequence(a, convert)
Type(b 2)
"rna"
Type( soapObject 2 ) SOAP services for information.
Type( s_dbtable sql column ) sarray with SQL column types of the database table s_dbtable.molcart, Name molcart
- returns the array with all elements but one thrown away from the groups of successive equal elements.
Unique ( sorted_array ) Unique ( unsorted_I_or_S_array [ sort | number ] ) number option).
Unique(.. sort ) and Unique(.. number) arrays can be combined into a table (see example below).
This table is similar to the [2,N] matrix returned by the Histogram function.
ii = {3 1 3 2 2 5 2 1} # let us form a table with values and frequencies
add column t Unique(ii sort) Unique(ii number)
show t
- returns the table made unique by one of its columns.
Unique ( table s_columnName )
Examples:
Unique( {1 1 2 3 3 3 4} ) # returns { 1 2 3 4 }
Unique( {1 1 2 2 1 1} ) # returns { 1 2 1 }Sort
- returns the Unix ( s_unix_command ) string
output of the specified unix command. This output is also copied to the s_out
string. This function is quite similar to the sys or unix
command. However the function, as opposed to the command, can be used in an expression and
be nested.
Examples:
show Unix("which netscape") # equivalent to 'unix which netscape'
#
if ( Nof(Unix("ls"),"\n") <= 1 ) print "Directory is empty"
Sarray( s_filePattern directory [ all ] )
set directory command
sys or unix command (e.g. unix ls )
- returns Value ( vs_var [R_referenceValues] ) rarray of selected variables. Both v_ (free) and V_ (all) variable selections can be used. The function considers variables only in the current object.
If the real array of reference values is provided then the torsion angles are returned in a 360. period closer to the reference values.
Examples:
build string "ASD"; minimize # a short peptide
ang=Value( v_//phi,psi ) # array of 4 phi-psi values
hbondlens=Value( V_//bh* ) # array of lengths for all H-X bonds
Value(v_//phi,psi {180.,180.,180.,180.}) # will convert -150. to 210.
SOAP services ).
Value ( soapMessage ) SOAP services for more information.
- returns Vector ( R_vector1 R_vector2 ) rarray [1:3], which is the vector product with components
{ v1[2]*v2[3] - v2[2]*v1[3], v1[3]*v2[1] - v2[3]*v1[1], v1[1]*v2[2] - v2[1]*v1[2] } Sum( R_n_vector1 R_n_vector2 )
- transforms an augmented affine 4x4 space transformation Vector ( M_matrix ) matrix into a
transformation vector.
Augment( ) function.
- returns i-th latent vector of the PLS model
Vector ( PLS_model i_num )Nof latent learn predict
- returns Version ( session ) string with the FlexLM license information
Version(session)
Files searched:/home/don/icmd/license.dat;/usr/local/flexlm/licenses/license.dat;*.lic Version ( [ full | number ] ) string containing the current ICM version.
The second field in the string specifies the operating system: "UNIX" or "WIN".
At the end there is a list of one-letter specifications of the licensed modules
separated by spaces (e.g. " G B R " ).
Option number just has the version itself (e.g. 3.1-03a )
full adds a few fields:
[Mar 18 2002 14:48]
Version ( graphics [ full ] ) string containing the OpenGL graphics driver vendor information.
With the full option the output is more verbose and lists the supported OpenGL extensions
and a number of driver-specific limitations, like the maximum number of light sources
supported.
See also: show version.
Example:
show Version( ) # it returns a string
if (Real(Version( )) < 2.6) print "YOUR VERSION IS TOO OLD"
if (Field(Version( ),2) == "UNIX") unix rm tm.dat
if (Field(Version( ),2) == "WIN") unix del C:\tm\tm.dat
if Version() == " D " print " Info> the Docking module license is ok"
show Version( number ) # returned 3.1-03 today
Version( graphic full )
GL_VENDOR = NVIDIA Corporation
GL_RENDERER = GeForce 6600 GT/PCI/SSE2
GL_VERSION = 2.0.1
GL_EXTENSIONS = GL_ARB_color_buffer_float ... WGL_EXT_swap_control
GL_MAX_VIEWPORT_DIMS = 4096 x 4096
GL_MAX_LIGHTS = 8
GL_MAX_CLIP_PLANES = 6
Version ( s_binaryFile [ binary | gui ] ) string version of the binary file ( binary option ) or
the version of the ICM executable used to save the file ( gui option ).
Version("tmp.icb" gui) # returns string
3.025j
if( Integer(Version("tmp.icb" binary ))> 6 ) print "OK"
- returns Volume ( grob ) real volume of a solid graphics object
(would not work on dotted or chicken wire grobs). ICM uses the Gauss theorem for
calculate the volume confined by a closed surface:
V = 1/3 * Sum( A * n * R )
where A is a surface area of a triangle, n a normal vector, and R is
is a vector from an arbitrary origin to any vertex of the triangle. It is
important that the grob is closed, otherwise, strictly speaking, the volume
is not defined. However, small surface defects will not affect substantially
the calculation. ICM minimizes possible error by rational choice of the origin,
which is mathematically unimportant for an ideal case. To define directions of
the normals the program either takes the explicit normals (i.e. they may be present
in an input Wavefront file) or uses the order of points in a triangle.
ICM-generated grobs created by the
make grob [ potential | matrix | map]
command have the correct vertex order (the corkscrew rule), while the
make grob skin
command calculates explicit normals.
The best way to make sure that everything is all right is to
display grob solid
and check the lighting. If a grob is lighted from the outside, the normals point
outwords, and the grob volume will be positive. If a grob is lighted from the inside
(as for cavities), the normals point inwords and the volume will be negative. If the
lighting, and therefore normals, are inconsistent you are in trouble, since Mr Gauss
will be seriously disappointed, but he will issue a fair warning from the grave.
The surface area is calculated free of charge and is stored in r_out . The surface
defect fraction, a relative area of the missing triangles, is returned in r_2out .
Normally this fraction should be zero.
Example:
read grob "swissCheese"
# divide one grob it into several grobs
split g_swissCheese
for i=3,Nof(grob)
# see, all the holes have negative volume
print "CAVITY" i, Volume(g_swissCheese$i)
endfor
See also: the Area( grob )} function, the split command and
How to display and characterize protein cavities
section.
- returns Volume ( r_radius ) real volume of a sphere, (4/3)Pi*R3
- returns Volume ( s_aminoAcids ) real total van der Waals volume of specified amino-acids.
- returns Volume ( R_a_b_c_alpha_beta_gamma ) real volume of a cell with the crystallographic unit cell parameters parameters {a,b,c} for a parallelepiped or
{a, b, c, alpha, beta, gamma} in a general case.
Volume ( R6_xyzXYZ box ) Box function), e.g.
read pdb "1xbb"
Volume( Box( a_2 ) box )
- returns Volume ( g_grob ) real volume confined by a grob.
Examples:
vol=Volume(1.) # 4*Pi/3 volume of unit sphere
vol=Volume("APPGGASDDDEWQSSR") # van der Waals volume of the sequence
vol=Volume({2.3,2.,5.,80.,90.,40.}) # volume of an oblique cell
- returns View( [ window ] ) rarray of 37 parameters of the graphics window and view attributes.
Some facts:
Screen coordinates fit into [-1,1] box.
(Note: Versions before 2010 returned 36 parameters, however the new version understands the old format as well).
With the window option the function returns only WindowWidth and WindowHeight .
# display something
V = View()
M1 = Matrix(V[ 1:16], 4)
M2 = Matrix(V[17:32], 4)
# Now any physical coordinate can be transformed into a screen coordinate:
XX = Augment( Xyz(a_//c* ) ) # function Augment adds the needed 4th coordinate.
show XX * M1 * M2 # atoms with a screen coordinate outside [-1,1] will not be visiblerotate view Rot( vector angle ) command. For example:
or use rotateView macro
rotate view Rot({1. 0. 0.} 90.) # for screen X coordinate
Example (how to save the image with 3 times larger resolution):
nice "1crn" # resize window
write image window=2*View(window) # 2-times larger image
returns View( "x" | "y" | "z" )rarray of 3 components of the screen X, Y, or Z axis. The vectors are normalized.
Example:
read pdb "1crn"
display a_ # rotate to the desired view
arrx = Grob( "ARROW" 10.*View("x") ) # make a 10A arrow
arry = Grob( "ARROW" 10.*View("y") ) # make a 10A arrow
display arrx,arry View( slide )rarray of the viewpoint parameters extracted from the slide .
View ( R_37_FromView, R_37_ToView, r_factor )
- returns rarray of the interpolated view
between the from and to view at the intermediate point 0≤ r_factor≤1.
If r_factor is out of the [0,1] range, the operation becomes extrapolation
and should be used with caution.
The camera view is changed in such a manner that the physical
space is not distorted (the principal rotation is determined
and interpolated, as are translation and zoom)
Example:
See also
nice "1crn" # manually rotate and zoom
r1= View() # save the current view
# INTERACTIVELY CREATE ANOTHER VIEW
r2= View() # save the new view
for i=1,100 # INTERPOLATION
set view View(r1,r2,i*0.01)
endforView (),
set view and
set view.
- returns View ( { "x"|"X"|"y"|"Y"|"z"|"Z" } ) rarray of 3 coordinates of the specified axis of the screen coordinate system.
Example:
build string "se ala"
display # rotate it now
show View("x")
g1=Grob("ARROW",3.*View("x"))
display g1
- returns Warning() logical yes if there was an warning in a previous command
(not necessarily in the last one).
After this call the internal warning flag is reinstalled to no.
- returns Warning ( string ) string with the last warning message.
In contrast to the logical Warning() function,
here the internal warning code is not reinstalled to 0, so that you can
use it in expressions like if Warning() print Warning(string) .
Example:
read pdb "2ins" # has many warnings
if Warning() s_mess = Warning(string) # the LAST warning only
print s_mess
Xyz ( as | grob , [ {Default X,Y,Z} ] ) Xyz ( as residue )
- returns matrix [ number_of_selected_atoms , 3]
in which each row contains x,y,z of the selected atoms.
Option residue will return the mean coordinates of groups of selected atoms in each selected residue.
Examples:
coord=Xyz(a_//ca) # matrix of Ca-coordinates
show coord[i] # 3-vector x,y,z of i-th atom
show Mean(Xyz(a_//ca)) # show the centroid of Ca-atoms
Xyz( as ring )matrix [ number_of_rings_in_selection , 3]
in which each row contains x,y,z of an ring from the selection
Xyz ( as r_interPointDistance surface )
- returns a subset of representative points at the accessible surface which are
spaced out at approximately r_interPointDistance distance. This distance from the
van der Waals surface (or skin ) is controlled by the vwExpand parameter.
Example:
build string "ala his trp"
vwExpand = 3.
mxyz = Xyz( a_ 5. surface )
display skin white
dsXyz mxyz
color a_dots. red
Xyz( M_abs_xyz, R_3_6cell|obs ) → M_fract_xyz Xyz( M_fract_xyz, R_3_6cell|obs, cell ) → M_abs_xyz
Xyz(Matrix({1. 2. 3.}),{10. 10. 10.}) # to fractionals
#>M
Xyz(Matrix({1. 2. 3.}),{10. 10. 10.} cell) # to absolute coordinates
0.1 0.2 0.3 #>M 10. 20. 30. Xyz( M_xyz, R_12abs_transformation [reverse|cell|transform] ) → M_xyz1_transformed Xyz( M_xyz, i_symm_transformation, s_sym_group, R3_cell|R_6_cell [cell|translate]) → M_xyz_transformed Xyz( M_xyz, i_symm_transformation, s_sym_group, R_6cell//R_3center ) → M_xyz_transformed
translate )
translate is provided or if {x,y,z} vector is appended to the 6-membered cell array
The coordinates will be translated after the transformation to the cells around
{x,y,z} .
translate or {x,y,z} ,
accordingly) is also used by the transform command. The transform command does automated centering to
the center of mass with the translate option or static with the translate={x,y,z} option.
makeBioMT macro.
Xyz( as s_smiles ) → Xyz_match Xyz( R_3N_x1y1z1x2y2z3...> ) → <M_Nx3_xyz Xyz ( R_6cell axis ) → M_ABCvectorsAugment(R_6 ) function.
Example:
c6 = {10. 10. 10., 120. 120. 120.}
Xyz(c6,axis)[1] # A vector
Xyz(c6,axis)[2] # B vector
Xyz(c6,axis)[3] # C vector