
Copyright © 2026, Molsoft LLC
Mar 30 2026
Google Search:
Keyword Search:| Prev | ICM Language Reference Macros | Next |
[ AlignTwoSequences | CalcArea | CalcBindingEnergy | CalcDihedralAngle | CalcEnsembleAver | CalcMaps | CalcPairSeqIdsFromAli | CalcSeqSimilarity | CalcPepHelicity | CalcProtUnfoldingEnergy | CalcRmsd | calcSeqContent | ConvertObject | IcmCavityFinder | DsCellBox | FindSymNeighbors | DsChem | DsCustom | DsPropertySkin | CalcEnergyStrain | CalcRoc | chemSuper3D | icmPmfProfile | dsPrositePdb | DsRebel | DsSeqPdbOutput | dsSkinLabel | DsPocket | dsStackConf | dsVarLabels | ds3D | DsXyz | Find_related_sequences | findFuncMin | findFuncZero | MakeAxisArrow | modifyGroupSmiles | Morph2tz | nice | cool | homodel | loadEDS | loadEDSweb | makeIndexChemDb | makeIndexSwiss | MakePdbFromStereo | makeSimpleDockObj | makeSimpleModel | mkUniqPdbSequences | PlaceLigand | plot2DSeq | plotSeqDotMatrix | plotSeqDotMatrix2 | plotBestEnergies | plotFlexibility | plotCluster | plotMatrix | PlotRama | plotRose | plotSeqProperty | predictSeq | prepSwiss | printMatrix | printPostScript | printTorsions | RefineModel | regul | rdBlastOutput | rdSeqTab | remarkObj | searchPatternDb | searchPatternPdb | searchObjSegment | searchSeqDb | searchSeqPdb | searchSeqFullPdb | searchSeqProsite | searchSeqSwiss | Seticmff | setResLabel | sortSeqByLength | ParrayToMol | ParrayTo3D | Torsion Scan | Convert2Dto3D | Convert3Dto3D | MakePharma | IcmMacroShape | IcmPocketFinder | EvalSidechainFlex | OptimizeHbonds | MergePdb ]
Macros provide you with a great mechanism to create and develop your ICM environment and adjust it to your own needs (see also How do I customize my ICM environment. ). Very often a repeated series of ICM commands is used for dealing with routine tasks. It is wise not to retype all these commands each time, but rather to combine them into a bunch for submission as a single command. Several examples follow.
alignTwoSequences : interface macro to the `Align function |
alignTwoSequences seq_1 seq_2 s_aliName ("NewAlignment") s_comp_matrix ("") s_alignmentAlgorithm ("ZEGA") r_GapOpen (2.4) r_GapExtension (0.15) i_maxPenalizedGap (99)
A macro to provide the interface to the Align( seq1 seq2 .. ) function and allows one to change global parameters influencing its behavior. Arguments:
- two sequences
- s_alignmentName
- s_comp_matrix ("default"|"gonnet"|"blosum45"|"blosum50"|"blosum62"|"dna"|"hssp"|"ident")
- s_alignmentAlgorithm ("ZEGA"|"H-align")
- r_Gap_Open (2.4)
- r_Gap_Extension (0.15)
- i_maxPenalizedGap (99)
read sequence msf "azurins.msf" alignTwoSequences Azur_Alcde Azur_Alcfa "ali2" "blosum45" "H-align" 2.6 0.1 99
calcArea : calculating solvent accessible surface area |
calcArea as_
calculates solvent accessible area of each selection in multiple objects and stores it in a table. If a molecule is specified in a multi-molecular object, the surface area of an isolated molecule is calculated and other molecules are ignored. The area is reported in square Angstroms and the probe radius is assumed to be waterRadius .
Output: the macro creates table AREA . The empty comment field is added for user's future use. If the table exists, new rows are appended. Example:
read pdb "1crn"
read pdb "2crn"
calcArea a_*.1 # calculate area around 1st molecule in each object:
show AREA
#>T AREA
#>-Selection---Total_Area--Comment----
a_1crn.1 2975.9 ""
a_2crn.1 4681.2 ""
See also:
- show area surface as1 [ as2 ] [ waterRadius=.. ] : the main command used in this macro. This command gives larger flexibility since it allows definition of the second selection of the vicinity for the calculation.
- surface area
- waterRadius
calcBindingEnergy: estimates electrostatic, hydrophobic and entropic binding terms |
calcBindingEnergy ms_1 (a_1) ms_2 (a_2) s_terms ("el,sf,en")
evaluates energy of binding of two complexed molecules ms_1 and ms_2 s_terms for the given set of energy terms s_terms. This macro uses the boundary element algorithm to solve the Poisson equation. The parameters for this macro have been derived in the Schapira, M., Totrov, M., and Abagyan, R. (1999) paper.
Example:
read object s_icmhome+"complex" cool a_ calcBindingEnergy a_1 a_2 "el,sf,en"
calcDihedralAngle: calculate an angle between two planes in a molecule |
calcDihedralAngle as_plane1 as_plane2
calculates an angle between the two planes specified by two triplets of atoms, specified by the as_plane1 and as_plane2 selections
An example in which we measure an angle between planes of two histidines:
build string "ala his his" # we use another macro here display atom labels calcDihedralAngle a_/2/cg,nd1,cd2 a_/3/cg,nd1,cd2 Angle= 131.432612 deg. (in).
calcEnsembleAver: Boltzmann average the energies of the stack conformations |
calcEnsembleAver r_temperature (300.) s_parameter ("Value(v_/2/phi)")
a macro showing an example of how to calculate a Boltzmann-weighted average given a conformational stack of conformation representatives. The stack may be formed as a result of a Monte Carlo simulation or created manually. The s_parameter string contains any expression returning the parameter to be averaged (e.g. "Value(v_/2/phi)" or "Distance(a_/2/ca a_/4/ca)" ).
Example:
build string "ala his his" set vrestraint a_/* # impose rotamer probabilities mncallsMC = 5000 montecarlo # a stack is formed with energies calcEnsembleAver 300. "Value(v_/2/phi)"
calcMaps: calculate five energy maps and write them to files |
calcMaps s_fileNameRoot ("rec") R_box r_gridSize (0.5)
calculates five energy grid maps for the current object with the grid size r_gridSize in the 3D box volume defined by the R_box . The maps are saved to files with names s_fileNameRoot_gc.map s_fileNameRoot_gh.map etc. and are deleted upon return from the macro. Be careful with selecting a box. You may focus the box on the area of interest (e.g. Box( a_/55,66 , 7.) ). To use the maps read them in, rename to m_gc m_gh, etc. and set terms "gc,gh,ge,gb,gs" . If you determined the box interactively you may just use the Box () function without arguments (it returns the parameters of the graphical box).
Example:
read object s_icmhome+"crn"
calcMaps "crn" Box( a_/15 4. ) 0.6
read map "crn_ge"
rename m_crn_ge m_ge
display m_ge {1 2 3 0 4 5 6}
# the maps can be used in another session
See also: GRID.gcghExteriorPenalty
Calculating the matrix of pairwise percent identities between sequences of from a multiple sequence alignment. |
calcPairSeqIdsFromAli ali l_original (yes)
calculates a pairwise sequence identities and returns them in two forms: as a table of pairs and as a matrix. Note:This macro is now obsolete and since the same result can be achieved with the Distance function. Example
read alignment "aln" name="aln" n=Nof(aln) mids = 100*(Matrix(n,n,1.) - Distance(aln )) # the pairwise seq. identities t = Table( mids, Name(aln), Name(aln) ) # to convert the matrix into pairwise table t = Table( mids, index ) # a simpler version with i,j
Prerequisites, Input Arguments and Options
| parameter | explanation |
|---|---|
| ali_ | a multiple (or pairwise) sequence alignment |
| l_original | a parameter that determines if pairwise alignments are extracted from the multiple alignment as is ( no ), or pairs of sequences are realigned to avoid influences of other sequences ( yes ). We recomment to use the yes value. |
Algorithm
- each of the n&mult;(n-1)/2 , where
- sequence identity is calculated as the number of identical columns in the alignment divided by
Output
| variable name | explanation |
|---|---|
| seq_ids table | contains names of two sequences and sequence identity, .seq1 .seq2 , .ident |
| seq_ids_mat matrix | n x n matrix of pairwise identity values |
Example
read alignment s_icmhome+"sh3.ali" calcPairSeqIdsFromAli sh3 yes show M_out show T_out
See also:
calcSeqSimilarity calculate sequence similarity according to an alignment |
calcSeqSimilarity aln s_refSeqName|i_refSeqPosInAli l_selection (no)
this macro returns a table, SeqSimilarity, with sequence identities with respect to the specified reference sequence ( s_refSeqName or i_refSeqPosInAli ). If a block of positions is
selected (e.g. by selecting the binding site, or directly in the alignment editor), additional columns will show sequence similarity in the selected positions.
calcPepHelicity: calculate average helicity of a peptide from trajectory frames |
calcPepHelicity s_movieName r_temperature (300.)
a macro showing an example of how to calculate the helicity of a peptide structure given an ICM trajectory file of the conformations accepted during a Monte Carlo run. A simulation using montecarlo trajectory option is a prerequisite for this macro. A good script prototype can be found in the $ICMHOME/_folding file. The trajectory option saves each accepted conformation to a trajectory file. The secondary structure of all transient conformations is assigned with the assign sstructure command.
Example:
% _folding # run the _folding script with the trajectory option. % icm read object "mypep" # the name of your peptide object calcPepHelicity "mypep" 600.
See also macro calcEnsembleAver
calcProtUnfoldingEnergy: rough estimate of solvation energy change upon unfolding |
calcProtUnfoldingEnergy ms ( a_1 ) i_mncalls ( 100 )
calculates an octanol/water transfer solvation energy for the given # conformation as compared to an extended chain conformation.
calcRmsd: calculate three types of Rmsd between protein conformations |
calcRmsd rs_1 (a_1/*) rs_2 (a_1/*)
calculates Ca-atom, backbone-atom, and heavy-atom RMSD for two input residue selections. The main effort in this macro is to take the internal symmetry of amino-acid sidechains into account.
For example, two phenylalanines related by the 180 degrees rotation of the xi2 angle are identical, but will have a non-zero Rmsd(a_1./phe a_2./phe) because cd1 and ce1 of one selection lay on top of cd2 and ce2 atoms of the second selection, respectively. To calculate this Rmsd correctly, we need to find the rotation The following residues have internal symmetry (or pseudo-symmetry): leu,tyr,phe,asp,glu,arg,val.
calcSeqContent |
calcSeqContent S_sequenceNames
analyzes amino acid composition of the input sequence or sequences. Specify quoted sequence name, pattern (e.g. "*_HUMAN" ) or "*" for all sequences.
Example:
read sequence s_icmhome+"seqs" calcSeqContent "*" # matches names of all sequences .. Statistics for 3 sequence(s): Azur_Alcde Azur_Alcfa Azur_Alcsp AA N % Expected A 42 10.34 7.85 C 9 2.22 2.55 ... calcSeqContent "*de" # sequences ending with 'de' Statistics for 1 sequence(s): Azur_Alcde Res N % Expected A 20 13.42 7.85 C 3 2.01 2.55 D 8 5.37 5.17 E 6 4.03 6.95
The columns are as follows:
- One-letter amino-acid code
- The total occurrence of the amino acid
- Relative percentage occurrence in the given set of sequences
- Expected mean occurrence of the amino acid in proteins
convertObject : creating a full atom model with types and charges from the source PDB file |
convertObject ms (a_) l_delete_water (yes) l_optimize_hydrogens (no) l_replace_the_original (no) l_display (no) l_optimize_his (yes) l_maskMissing (no) l_assignHeteroCharges (yes) s_options ("") auto
This macro converts a non-ICM object (e.g. PDB structure) into and ICM object and performs some additional refinements. Input: a selection of molecules (e.g. a_b,c ) and the following arguments:
- l_delete_water (yes) : automatically deletes all waters
- l_optimize_hydrogens ( no ) : performs global optimization of hydrogens for best hydrogen bonding network
- l_replace_the_original ( no ) : keeps the same name as the original object and deletes it
- l_display ( no ) : displays the converted object
- l_optimize_his (--yes) : His protonations states will be tried, An and Gln flips will be tried and Cys in the the vicinity of Zn, Cu, Fe and Co to cym
- l_maskMissing (no) : ICM will always add missing atoms for known residues. When l_maskMissing = yes these atoms will be masked off (hidden). They will be excluded from simulations, maps for docking etc.. This is for side-chains only it will not fill gaps in the backbone.
- l_assignHeteroCharges (yes) : will set ligand charged state according to Molsoft's pKa model.
There are also some string options
For example if you are reading in a protein structure prepared in a different software and you do not need to add hydrogens and optimize or make any changes you can use:
convertObject a_ no no no no no no no
or if you want to convert to an ICM object, and optimize hydrogens, His, Asn, Gln and add missing side-chain atoms and keep tight waters and optimize ligand tautomers you could use:
convertObject a_ no yes no no yes no no "water=tight tautomer"
The procedure will do the following :
Output:
The macro returns r_residualRmsd value containing the Rmsd of the model atoms from the
equivalent template atoms (the same value is returned by the convert command in r_out ).
If this residual is greater than 0.5 , it usually means some problems with the
conversion (e.g. unusual residues, missing parts, etc.).
This macro can be found in the __macro file or viewed using the edit command. It calls two other macros:
Example:
Prerequisites and Arguments
The algorithm
The output
Example:
calcRoc R_scores L_01labels r_score_error (0.) l_linear_auc (no) l_keep_table (no) l_showresults (yes)
compute the normalized measure of the recognition strength of the provided score ( R_scores )
given a matching array of zero and one labels for 'noise' and 'signal' respectively.
Lower scores are considered better (comes from energy), invert the sign of the scores otherwise.
Example: scores { 2.1 4. 3. 44.} and labels {1 0 0 0} correspond to a perfect recognition since 2.1 is the lowest score.
The normalized measure returns the value of 100. for any perfect separation of signal from noise and values close to zero
for a random subset of noise. The normalized measure is based on the area under curve in the following
axis: the number of signal records in the top N scores versus the record rank (or the square root of it).
The rank is defined as the order in a sorted array. The normalized measure is the following:
AUC..normalized = 100*( AUC - AUCrandom )/(AUCperfect / AUCrandom )
Input
Algorithm
OutputIf the l_keep_table flag is set to no, the four values are returned as R_out[1:4].
Example:
chemSuper3D os_in os_template l_optimize (no) l_display (no) l_all (no) auto
performs a sequential flexible superposition of multiple molecules in the os_in selection on a single rigid template os_template .
For static superposition, use superimpose chemical .
Prerequisites and Input
Parameters and options
Output
See also:
See also:
dsPocket ms_ligand (a_H [1]) s_GrobName ("") l_overwrite (yes) l_ds_xstick_hb_labels (no)
dsPocketRec ms_ligand (a_H [1]) ms_receptor (a_!H ) r_margin (6.5) s_GrobName ("") l_overwrite (yes) l_ds_xstick_hb_labels (no)
display the receptor pocket around the selected ligand ms_ligand.
Only the largest contiguous pocket surrounding the ligand is retained
for clarity. Macro also colors the molecular surface
by hydrogen bonding potential and hydrophobicity if receptor is as ICM object.
Best used with the ligand shown in cpk, if the ligand is small.
The first macro ( dsPocket ) is trying to guess what the selecting of atoms for the binding pocket is. Then it calls dsPocketRec
The second macro ( dsPocketRec ) has identical arguments plus explicit receptor selection.
The dsPocket guess about the receptor:This is the order of guesses about the selection of the binding pocket molecules:
Prerequisites and arugments of the dsPocketRec macro
The procedure
Output
This macro replaces dsSkinPocket and dsSkinPocketIcm macros.
find_related_sequences auto ms (a_*.A)
identifies pairs of molecules (chains) with similar sequences in the input selection.
(see also Select( seq .. ) )
Input and arguments
* for each pair of amino-acid chains in all objects the following:
Output
Example:
findFuncZero s_Function_of_x ("Exp(-Exp(-x))-0.5") r_xMin (0.) r_xMax (1.) r_eps (0.00001)
makeAxisArrow rs ( a_/10:18 ) i_length (10) r_radius ( 0.12 ) r_head_width_ratio ( 2. )
this macro determines the direction make an arrow with specified parameters.
Input and arguments:
modifyGroupSmiles as_group s_Group l_reset_MMFF_types (yes) l_reassign_MMFF_charges (yes) l_optimize_geometry (no) auto
attaches a chemical group to the specified anchor atom.
Input and Arguments
Output
morph2tz rs_loop ( a_/ ) i_nIter l_store_in_object (no) l_play_morph (yes)
this macro generates a stack of intermediate conformations for an fragment in an object.
This intermediate conformations are obtained by a restrained minimization using the source
and target structures, as well as the steric restrains.
Prerequisites and arguments
The algorithm
The output
loadEDS s_pdb ("1mui") r_sigma (0.)
read the 2fo-fc map file with crystallographic electron density from one of three sources. (for fo-fc map use loadEDSweb )
The map named m_s_pdb (e.g. m_1crn ) is loaded from the Uppsala server or a local directory.
If the macro ends up loading it from the web, it writes the map a local repository as s_pdb.map file
for fast future access.
To read the map file three locations are checked in the following order:
The arguments
The procedure
The output
New files, if the map was downloaded from the web.
loadEDSweb auto s_pdbcode ("1mui") [ s_maptype ("2fofc") ]
downloads 2fofc (or other s_maptype) map from the Uppsala electron density server "http://eds.bmc.uu.se/"Creates a map object m_s_pdbcode . This macro can be used to load Fo-Fc maps ( s_maptype "fofc" )
This macro is used by the loadEDS macro.
Example:
makePdbFromStereo R_xl R_xr R_yl R_yr r_stereoAngle ( 6. )
makeSimpleDockObj os s_newObjectName
This macro builds an ICM object from simplified residues described in the
residue library. The goal is to convert an all-atom molecular object into an
object in simplified representation for fast docking calculations.
makeSimpleModel seq ali os
This macro rapidly builds a model by homology using simplified residues described in the
residue library. Input data are the sequence of the model, seq_ and
alignment ali_ of the model's sequence with
the template object os_ .
placeLigand ms_movable_ligand_ICM as_static_target_ICM r_effort (1.) l_display l_debug
placeLigand docks a ligand molecule in a single ICM object to the specified atom selection in ICM-converted objects.
Selections of groups/ligand in multiple ICM Objects is allowed as a destination.
If additional restraints/tethers are needed, use distance tool to define them.
Tethers that are set directly via 'set tether' will be respected too.
The macro saves a stack of preferred superposisions. No additiona entities/objects/maps are created.
Parameters:
Example:
generates a residue table and three interactive plots for non-glycines,
glycines and omega angles (see Tools/Analysis menu)
plotRamaEps rs l_show_residue_label (no) l_shaded_boundaries (yes)
predictSeq s_seq ("1crn_m") s_fileName ("plot") l_predictSstr (no) i_numPerLine (100)
regul rs (a_!W) s_ngroup ("nh3+") s_cgroup ("coo-") l_delete_water (yes) l_shortOutput (yes)
creates a regularized
ICM-model of an input residue selection ( rs_ ) modified by the N- and C-terminal
groups ( s_ngroup and s_cgroup, empty "" strings are
allowed);
searchSeqFullPdb s_projName ("pdb1") r_probability (0.01) l_appendProj (no)
sequence search of all currently loaded sequences through all proteins from the collection
s_pdbDir+"/derived_data/pdb_seqres.txt.Z", a subset of PDB sequences with given degree of
mutual dissimilarity. Found hits and their specs
are collected in the output table file s_projName.If logical flag l_appendProj is on data will be appended
to the existing table. Similarity of hits to the query sequence(s) is controlled by
parameter r_probability (see
Probability()).
searchSeqProsite seq
compares input sequence against all sequence patterns collected in the
PROSITE database.
Examples:
find pattern,
find database pattern=s_pattrn,
find prosite.
set_icmff [ r_vwSoftMaxEnergy (4.) ]
sets parameters and load residue and vw-parameter libraries necessary for the icmff force field.
You can redefine the softness (a.k.a. vw-truncation limit) with an optional second argument.
Note that the new libraries have a bond planar angle at the Ca atom unfixed.
Any new peptide build after this command will have icmff-compatible residues. If you already a PDB
object, conversion will create an icmff-compatible object. If your object has already been converted before
the set_icmff command, it needs to be stipped and converted again.
E.g.
Reference:
Arnautova YA, Abagyan RA, Totrov M.
Development of a new physics-based internal coordinate mechanics force field and its application to protein loop modeling.
Proteins. 2011 Feb;79(2):477-98.
The actual macro contains the following:
sortSeqByLength
sort sequences by their length and suggest outliers.
parrayToMol P_m
This macro converts each elements of chemarray to a 3D object preserving coordinates
Example:
See also: parrayTo3D convert2Dto3D
parrayTo3D P_m
This macro converts each elements of chemarray to a 3D object optimizing geometry.
Example:
See also: parrayToMol convert2Dto3D
torScan vs_oneTorsionSel r_step_in_deg (5.) l_optimizePolarH (no)
Input and prerequisites:
Arguments and Options:
Output:
Example:
convert2Dto3D os l_build_hydrogens (yes) l_fixOmegas (yes) l_display (no) l_overwrite (yes)
the main macro that is used to convert 2D molecules from an .sdf or .mol file to a full atom optimized ICM 3D object.
It is used by many ICM tools and scripts (multiple superpositions, conformational generator, docking etc.).
Prerequisites and arguments
Output
See also: parrayTo3D parrayToMol, convert3Dto3D (coordinate-preserving conversion of a 3D mol file)
convert3Dto3D os l_build_hydrogens (yes) l_display (no) l_overwrite (yes)
same as convert2Dto3D but the x,y,z coordinates are preserved.
makePharma as_obj s_name ("pharm") l_points (yes) l_display (yes) auto
This macro creates a pharmacophore object from as_obj. s_name will be used for the result.
if l_points is yes pharmacophore will be created (see show pharmacophore type) otherwise input molecule will
be split by rotation bonds.
See also: find pharmacophore , pharmacophore
icmMacroShape as i_complexity (8) r_gridStep (0.) r_contourLevel (1.2) l_colorByDepth (yes) l_fast (no) l_display (no) s_rainbow ("blue/white/red")
The icmMacroShape macro generates a low resolution surface
Prerequisites and arguments.
The algorithms follows these steps:
The output
icmPocketFinder as_receptorMol r_threshold ( 4.6 ) l_displayPocket (yes) l_assignSites (no)
Use this macro to find binding pocket(s) for the input as_receptorMol.
Arguments:
The macro creates a table which contains a list of all pockets found. The table is sorted by pocket volume.
Example:
evalSidechainFlex rs_residues (a_) r_Temperature (600.) l_atomRmsd (yes) l_color (yes) l_bfactor (no) l_entropyBfactor (no)
evalSidechainFlex systematically samples rotamers for each residue side-chain in the input selection
and uses resulting conformational ensembles to evaluate energy-weighted RMSDs for every side-chain atom.
These are stored in the 'field' values on atoms and can be used for example to color the structure by side-chain flexibility.
Conformational entropy for each residue side-chain is also calculated and stored in a table ATOM_FLEX
Arguments:
optimizeHbonds as l_rotatable_hydrogens (yes) l_optimizeHisAsnGln (yes)
optimizeHbonds optimize the hydrogen bond network of the selected atom.
Arguments:
mergePdb rs_source ( a_1.1/20:25 ) rs_graft ( a_2.1/20:25 ) s_combo ("combo")
mergePdb combines two PDB object into one and assign continuous residue number to the combined object.
Arguments:
Selection tags ( Select ( as tag ) returns the selection ) :
read pdb "1abe"
convertObject a_1,2 yes yes yes no
show r_residualRmsd
icmCavityFinder: analyze and display cavities
icmCavityFinder as ( a_A ) r_minVolume ( 3. )
calculates and displays cavities in a molecular structure.
These cavities are sorted by size, and displayed.
To display the transparent outer shell edit the macro and activate this feature.
The r_minVolume parameters defines the volume of the smallest retained cavity.
Increase it if you want only large cavities.
For each cavity this macro calculates volume V (in square Angstroms),
area A and an effective radius R (compare it with the radius of
a water molecule of 1.4A).
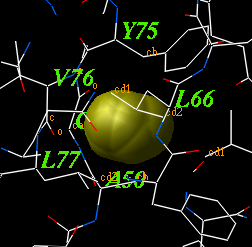
The icmCavityFinder macro uses two powerful features of ICM-shell:
Example:
read object s_icmhome+"1qoc"
delete a_w* # remove water molecules
icmCavityFinder a_1 yes 4.
3
dsCellBox: displays crystallographic unit cell
dsCellBox os (a_)
displays unit crystal cell box for the specified object os_ generated
according to crystal symmetry parameters.
This tiny macro extracts the cell from the object using the
Cell function and makes a grob out of this array with the
Grob function.
macro dsCellBox os_ (a_)
gCell = Grob ("cell" Cell(os_))
display gCell magenta
keep gCell
endmacro
See also: findSymNeighbors
findSymNeighbors: cell and crystallographic neighbors
findSymNeighbors as ( as_graph ) r_radius (7.) l_append (no) i_extend_by (2) l_keepEntireChain (no) l_display (yes)
finds and builds symmetry related molecules around the input selection.
The symmetry related atoms are generated according to the crystallographic symmetry group and cell dimensions.
read pdb "2ins"
findSymNeighbors a_1,2 5. no 2 no no # a_2insSym. object with 14 fragments is created.
show nSymNeighbors, S_neighborAtoms
7
>S S_neighborAtoms
a_2ins.1:2,5/*|a_2ins.c/^N21|a_2ins.d/^G8:^S9,^V12:^E13,^Y16,^G20:^K29
a_2ins.5/*|a_2ins.b/^S9:^H10|a_2ins.c/^L13|a_2ins.d/^V2:^Q4,^L6,^A14,^Y16:^E21
a_2ins.b/^K29
a_2ins.5/*|a_2ins.b/^H10
a_2ins.c/^G1,^E4|a_2ins.d/^P28:^A30
a_2ins.b/^R22
a_2ins.c/^A8:^V10|a_2ins.d/^N3,^H5
a_2ins.c/^G1,^N18:^Y19,^N21|a_2ins.d/^F25,^K29:^A30
dsChem : chemical style display
dsChem as (a_)
3D display of the input atom selection in chemical
style and on white background.
If you want to 'flatten' the molecule you can perform
a procedure from the following example:
build string "trp" # you need an ICM object
tzMethod = "z_only" # tether to the z-plane
set tether a_ # each atom is tethered to z=0
minimize "tz" # keep the cov. geometry
dsCustom: extended display and property-coloring
dsCustom as (a_) s_dsMode ("wire") s_colorBy ("atom") l_color_only (no)
Displays the specified representation
( "wire", "cpk", "ball", "stick", "xstick", "surface", "ribbon" )
of a molecular selection and colors the selection according to the
following series of features:
dsPropertySkin: display molecular surfaces colored by properties essential for binding
dsPropertySkin as_sel (a_) l_wire (yes) l_biggestBlobOnly (no)
displays essential properties of molecular surfaces which are essential for binding
small ligands, peptides or other proteins.
The first argument is a selection of atoms involved in the surface calculation. The second
argument allows you to display the surface as:
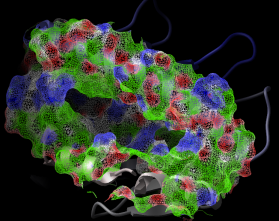
The color code:
Example shown:
read pdb "1a9e"
delete a_w*
convert # convert to ICM for map calculations
# select receptor atoms 9. away from the peptide with Sphere
cool a_ # display ribbon
dsPropertySkin Sphere( a_3 a_1 9. ) yes no
# adjust clipping planes for better effect
write image png
Interactive surface display under GUI The same can be performed interactively on ICM objects with the popup-menu:
calcEnergyStrain: analyzing energy strain in proteins
calcEnergyStrain rs ( a_/A ) l_colorByEnergy (yes) r_max (7.)
calculates relative energy of each residue
for residue selection rs_ ; and colors the selected residues
by strain ( if logical l_colorByEnergy is "yes" ).
The r_max argument determines the range represented by the color
gradient (i.e. residues strained beyond 7. will still be shown in red).
This macro uses statistics obtained in the Maiorov, Abagyan, 1998 paper.
Example:
read object s_icmhome + "crn" # an ICM object
calcEnergyStrain a_/A yes 7.
show ENERGY_STRAIN
Calculate normalized 'area under curve' for the hit enrichment histogram vs rank or square root of hit rank
Otherwise a table called tabauc is created.
N=1000; n=100
calc_nosauc Random(0.,1.,n,"gauss")//Random(3.,1.,N-n,"gauss") Iarray(n,1)//Iarray(N-n) 0.2 yes no
show R_out
show tabauc.nosauc, tabauc.nmr
# select columns X and N and plot the enrichment curve.
# play with the parameters
chemSuper3D
icmPmfProfile
icmPmfProfile os ( a_ ) l_accessibilityCorrection (yes) l_display ( no )
calculates a statistical energy of mean-force for each residue of a provided object.
This energy is calculated with the "mf" parameters defined in the icm.pmf
file. The residue energies are then normalized to the expected mean and standard
deviation of the same residue in real high resolution structures.
The mean energy value can be calculated as a function of its solvent accessibility
if l_accessibilityCorrection is set to yes.
The calculated table contains residue energies and accessibilities. These values
can be used to color residues of the molecule according to those values.
In an example shown here we build a model (using the build model command
of HCV protein on the basis of another viral coat protein. Then the profile
was calculated for the model and the original structure. The calculation clearly
shows the problematic regions of the model (the red parts) while the source
structure looks quite reasonable.
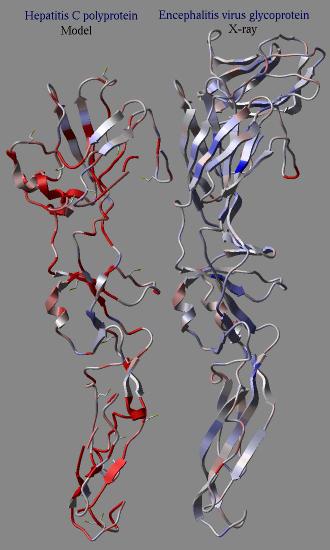
dsPrositePdb
dsPrositePdb ms (a_*) r_prositeScoreThreshold (0.7) l_reDisplay (no) l_dsResLabels (yes)
Finds all PROSITE pattern-related fragments in the current object and
displays/colors the found fragments and residue labels.
dsRebel: surface electrostatic potential
dsRebel as (a_*) l_assignSimpleCharges ( no ) l_display (no)
generates the skin representation of the molecular surface colored according
to the electrostatic potential calculated by the REBEL
method (hydrogen atoms are ignored).
The coloring is controlled by the maxColorPotential and TOOLS.rebelPatchSize parameter.
This macro uses a simplified charge scheme
and uses only the heavy atoms for the calculations for the sake of speed.
dsSeqPdbOutput : visualize the sequence similarity search results
dsSeqPdbOutput s_projName ("brku") l_resort (no)
Goes through a list of PDB hits resulting in
find database command
and displays alignment(s) of the input sequence(s) with the found PDB structures
and SWISSPROT annotations.
dsSkinLabel
dsSkinLabel rs (a_/*) s_color ("magenta")
For all residues specified by the input residue selector, rs_,
displays residue labels shifted toward the user to make the labels visible
when skin representation is used.
dsPocket and dsPocketRec
These macros can also be used to show protein-protein interface.
Example:
read object s_icmhome+"complex"
cool a_
dsPocket a_1 "" yes # shows the surface of a_1
dsStackConf
dsStackConf as (a_//n,ca,c) i_from (1) i_to (Nof(conf)) s_superimpRes ("*")
displays superimposed set of conformations from a conformational
stack for given selection as_.
dsVarLabels
dsVarLabels
displays color labels for different types of torsion variables.
ds3D
ds3D auto M_dist S_names ({""}) S_comments ({""})
display 3D coordinates corresponding to an input square distance data matrix.
Relative errors (in percent) of embedding to 3D space are in R_out: first entry
is for the total error, next three are for X, Y and Z coordinates.
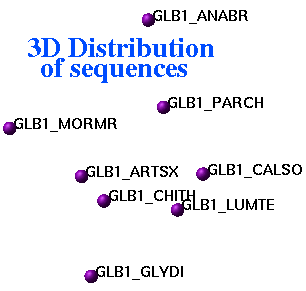
Representation of inter-sequence evolutionary distances in three-dimensional space
dsXyz : display
dsXyz M_3coor
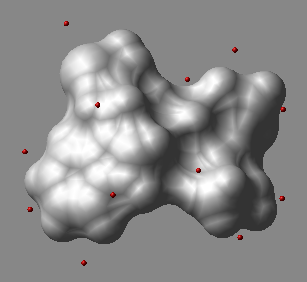 displays points from the N_atoms x 3 matrix of M_3coorin 3D space as blue balls.
The origin of the Nx3 matrix is not important.
The macro creates an object called a_dots.
In this object each dot is a one-atom residue called 'dot'. The atom type
is arbitrarily assigned to oxygen, and the atom names are 'o'.
displays points from the N_atoms x 3 matrix of M_3coorin 3D space as blue balls.
The origin of the Nx3 matrix is not important.
The macro creates an object called a_dots.
In this object each dot is a one-atom residue called 'dot'. The atom type
is arbitrarily assigned to oxygen, and the atom names are 'o'.
One can further manipulate this object, e.g. color a_/12:15/o green .
An example in which we generate sparse surface points at vwExpand distance around a molecule and display them.
build string "ala his trp"
mxyz = Xyz( a_ 5. surface )
display skin white
dsXyz mxyz
color a_dots. red
find_related_sequences macro: identify similar and superposable chains in multiple objects
The procedure
read pdb "1arb"
read pdb "1arc"
find_related_sequences a_*.*
show related_sequences
findFuncMin
findFuncMin s_Function_of_x ("Sin(x)x-1.") r_xMin (-1.) r_xMax (2.) r_eps (0.00001)
minimizes one-dimensional functions provided as a string with the function expression.
The macro uses successive subdivision method, and assumes that the function derivative
is smooth and has only one solution in the interval
Example:
findFuncMin "Sin(x)*x-1." , -1. 2. 0.00001
-1.000000 < x < 0.500000
-0.250000 < x < 0.500000
-0.062500 < x < 0.125000
....
-0.000004 < x < 0.000008
-0.000004 < x < 0.000002
findFuncZero
finds a root of the provided function of one variable with specified brackets with iterations.
E.g.
findFuncZero "x*x*x-3.*x*x" 1. 33. 0.00001
-> x=17.000000 F=4046.000000
-> x=9.000000 F=486.000000
-> x=5.000000 F=50.000000
-> x=3.000000 F=0.000000
Determining selection axis and making an arrow.
Output
Example:
read pdb "1crn"
display ribbon
makeAxisArrow a_/23:30 10 0.2 2.1
color axis_m blue
modifyGroupSmiles
morph2tz : generating sterically plausible intermediate conformation for conformational morphing
nice
nice auto s_PdbSelection ("1crn") l_invert (no) l_wormStyle (no) l_append (no) l_nodisplay (no)
reads and displays a PDB structure in ribbon representation; colors each
molecule of the structure by colors smoothly changing from blue (at N-terminus)
to red (at C-terminus).
The auto keyword allows one to skip N last arguments.
Example:
nice "365d" # new DNA drug prototype
nice "334d" # lexitropsin, derivative of netropsin
cool
cool auto rs (a_) l_static (no)
similar to the macro
nice
above, but refers to a residue selection.
homodel
homodel ali l_quick (yes)
homology modeling macro. The first sequence in the input alignment should contain
the sequence of a PDB template to which the modeling will be performed.
If flag l_quick is on, only an approximate geometrical model
building is performed.
You can also use the build model command directly.
loadEDS
The following ICM shell objects:
loadEDSweb
loadEDSweb "1mui" "2fofc"
display m_1mui
# or
loadEDSweb "1mui" "fofc"
makeIndexChemDb
makeIndexChemDb s_dbFile ("/data/acd/acd.mol") s_dbIndex ("/inx/ACD") s_dbType ("mol2") S_dbFields ({"ID"})
Creates and saves an index to a small compound database existing in
standard "mol" or "mol2" formats (specified by the s_dbType parameter).
s_dbIndex defines full-path root name of several index-related files.
String array S_dbFields specifies fields of
the input database which are indexed by the macro.
An example in which we index the cdi.sdf file and generate the cdi.inx file in
a different directory:
% icm
makeIndexChemDb "/data/chem/chemdiv/cdi.sdf" "/data/icm/inx/cdi" "mol" {"ID"}
makeIndexSwiss
makeIndexSwiss s_swiss (s_swissprotDat) s_indexName (s_inxDir+"SWISS.inx")
Creates and saves an index to the SWISSPROT sequence database
(datafile s_swiss).
s_indexName defines the root name of several index-related files
with respect to ICM user directory, s_userDir.
makePdbFromStereo: restore 3D coordinates from a stereo picture
transforms two stereo sets of two-dimensional coordinates in arbitrary scale into 3D coordinates.
See also: How to reconstruct a structure from a published stereo picture .
makeSimpleDockObj
makeSimpleModel
mkUniqPdbSequences
mkUniqPdbSequences auto i_identPercent ( 1 )
Creates a collection of PDB sequences with specified degree of mutual dissimilarity,
i_dentPercent.
placeLigand: flexible superposition of a ligand to atom selection
read object s_icmhome + "biotin.ob"
build smiles "CCCC(=O)[O-]" name="lig"
display xstick a_biotin,lig.
pause
placeLigand a_lig. a_1.1 3.
plot2DSeq
plot2DSeq ali_
generates a 2D representation of "distances" between each pair of sequences
from the input alignment.
plotSeqDotMatrix
plotSeqDotMatrix seq_1 seq_2 s_seqName1 ("Sequence1") s_seqName2 ("Sequence2") i_mi (5) i_mx (20)
generates an EPS file in which local sequence similarities between two sequences are shown
in the form of a two-dimensional dot-matrix plot.
Significance of local sequence similarities is shown by logarithm
of the probability values and is calculated in multiple windows from
i_mi to i_mx. The log-probability values are color-coded as follows: light blue: 0.7, red 1.0.
plotSeqDotMatrix2
plotSeqDotMatrix2 seq_1 seq_2 s_seqName1 ("Sequence1") s_seqName2 ("Sequence2") i_mi (5) i_mx (20)
generates an EPS file in which local sequence similarities
between two sequences are shown in the form of a two-dimensional dot-matrix plot.
Significance of local sequence similarities is shown by
( 1. - Probability(..) ) values and is calculated in multiple windows from
i_mi to i_mx. The ( 1 - P ) values are color-coded as follows: light blue: 0.7, red 0.99.
plotBestEnergies
plotBestEnergies s_McOutputFile ("f1,f2") r_energyWindow (50.) s_extraPlotArgs ("display")
plots profile of energy improvement during an ICM Monte Carlo simulation. Data
are taken from the MC output log file or files, s_McOutputFile.
You can specify a single output file (e.g. "f1.results"), or several files,
e.g. "f1.ou,f2.ou", or drop the default ".ou" extension, e.g. "f1,f2,f2".
This macro gives you an idea about the convergence between several runs.
plotFlexibility
plotFlexibility seq i_windowSize (7)
calculates and plots flexibility profile for input sequence seq_ and
smooths the profile with i_windowSize residue window.
plotCluster
plotCluster M_distances S_names ({""}) s_plotArgs ("CIRCLE display {\"Title\" \"X\" \"Y\"}")
plot distribution of clusters. Arguments:
for i=1,n-1
di[i,i]=0.
for j=i+1,n
# takes care of -179 and 181, base 360 is the default
dif=Remainder(v[i]-v[j])
# angular RMSD
di[i,j]=Sqrt(Mean(dif*dif))
di[j,i]=di[i,j]
endfor
endfor
di[n,n]=0.
plotMatrix
plotMatrix M_data s_longXstring S_titles ({"Title","X","Y"}) s_fileName ("tm.eps") i_numPerLine (10) i_orientation (1)
generates combined X-Y plot of several Ys (2nd, 3rd , etc. rows of the input
matrix M_data) versus the one X-coordinate, assumed to be the first row of
the matrix. i_numberPerLine parameter defines the size of the plotted block size
if the number of data points is greater then i_numberPerLine.
i_orientation equal to 1 defines portrait orientation of the output plot,
landscape otherwise.
plotRama and plotRamaEps
plotRama rs
generates a phi-psi Ramachandran plot of an rs_ residue selection.
If logical l_show_residue_label is on, the macro marks the residue labels.
If l_shaded_boundaries is on, the allowed (more exactly,
core)
regions are shown as shaded areas; otherwise the contours of the core regions
are drawn.
plotRose
plotRose i_prime (13) r_radius (1.)
just a nice example of a simple macro generating "rose" plot.
plotSeqProperty
plotSeqProperty R_property s_seqString S_3titles {"Y property","Position","Y"} s_fileName ("tm.eps") i_numPerLine (30) s_orientation ("portrait")
a generic macro to plot local sequence properties. Modify it for your convenience.
Here is an example in which we plot residue b-factors along with the crambin sequence.
s_seqString could be the sequence (e.g. String(1crn_m) ) or secondary structure,
(e.g. Sstructure(1crn_m) ) or any other string of the same length as the sequence.
read pdb "1crn"
make sequence
b = Bfactor( a_/* )
plotSeqProperty b String(1crn_m ) {"" "" ""} "tm.eps" 20 "portrait"
predictSeq
prepSwiss
prepSwiss s_IDpattern ("VPR_*") l_exclude (yes) s_file ("tm")
extracts all sequences from the SWISSPROT database which
exclude ( l_exclude= yes ) or include ( l_exclude= no) the
specified sequence pattern, s_IDpattern
and creates a set of database files with the rootname s_file
intended to use in the command
find database.
printMatrix
printMatrix s_format (" %4.1f") M_matrix (def)
prints matrix M_matrix according to the input format
s_format.
printPostScript
printPostScript s_ofPrinterName ("grants")
converts the current content of the graphics window to a PostScript file and
directs it to the s_ofPrinterName printer.
printTorsions
printTorsions rs (a_/A)
outputs all torsion angles of the input residue selection.
refineModel: globally optimize side-chains and anneal the backbone
refineModel i_nRegIter (5) l_sideChainRefinement (no)
This macro can be used to improve any ICM model. The model can come from
the build model command or the convert command or regul macro, etc.
It performs
To perform only the side-chain refinement, set the i_nRegIter argument to 0 .
regul
rdBlastOutput
rdBlastOutput S_giArray
reads a set of sequences defined in a BLAST's output file,
S_giArray from the NCBI database.
rdSeqTab
rdSeqTab s_dbase ("NCBI")
reads a set of sequences listed in the
ICM-table SR, an output
of
find database command,
from the database defined by s_dbase.
remarkObj
remarkObj
allows editing an annotation (comment) of the current object.
Existing comment (if any) is read in an editor and after modification assigned to the object.
searchPatternDb
searchPatternDb s_pattern ("?CCC?") s_dbase ("SWISS")
searches for the pattern in the sequences of the specified indexed
database s_dbase.
searchPatternPdb
searchPatternPdb s_pattern
searches for the specified pattern in pdb sequences taken from the foldbank.db file.
Example (first hydrophobic residue, then from 115 to 128 of any residues,
non-proline and alanine at the C-terminus):
searchPatternPdb "^[LIVAFM]?\{115,128\}[!P]A$"
searchObjSegment
searchObjSegment ms i_MinNofMatchingResidues (20) r_RMSD (5.)
for given molecule ms_ finds all examples of similar 3D motifs not
shorter than i_MinNofMatchingResidues residues with the accuracy
r_RMSD A in the ICM protein fold database.
searchSeqDb
searchSeqDb s_projName ("sw1") S_seqNames ({""}) r_probability (0.00001) l_appendProj (no) s_dbase ("SWISS")
search the database s_dbase using query sequence(s) specified in
S_seqNames. Found hits and their specs are collected in the output table file
s_projName.tab. If logical flag l_appendProj is on data will be appended
to the existing table. Similarity of hits to the query sequence(s) is controlled by
parameter r_probability (see Probability()).
searchSeqPdb
searchSeqPdb s_projName ("pdb1") r_probability (0.01) l_appendProj (no)
sequence search of all currently loaded sequences in the sequences of the proteins from the
fold bank collection. Found hits and their specs
are collected in the output table file s_projName.
If logical flag l_appendProj is on data will be appended
to the existing table. Similarity of hits to the query sequence(s) is controlled by
parameter r_probability (see Probability()).
searchSeqFullPdb
searchSeqProsite
read sequence "zincFing.seq" # load sequences
find prosite 2drp_d # search all < 1000 patterns
# through the sequence
find profile 2drp_d # search profile from prosite database
See also:
searchSeqSwiss
searchSeqSwiss seq_
Searches for homologs of the query sequence seq_
in the SWISSPROT database.
set_icmff setting the new ICMFF force field
read pdb "1crn"
set_icmff 5.
convertObject a_
# or
read binary "icmob.icb"
set_icmff
strip a_
convert # or convertObject
macro set_icmff auto r_vwSoftMaxEnergy (4.)
LIBRARY.res = {"icmff"}; ffMethod = "icmff"
vwSoftMaxEnergy = r_vwSoftMaxEnergy
read library energy
dielConst = 2.; electroMethod = "distance dependent"
set type "atomic" { 0.0080,0.0220,-0.0900,-0.2240,-0.1760,-0.0630,-0.0350,-0.2240,-0.0960,-0.1160,-0.0120,-0.0510,0.0080,0.0080,-0.0630,-0.0900,-0.0900,-0.1760,-0.0900,0.0,0.0100,0.0100,0.0100,0.0100,0.0100}
set term only "bb,vw,14,hb,el,to,sf"
vwMethod="soft"
flipStepPb=0.25
keep electroMethod dielConst ffMethod visitsAction vwMethod vwSoftMaxEnergy
endmacro
setResLabel
setResLabel
moves displayed atom labels to the atoms specific to each residue type.
sortSeqByLength
parrayToMol macro
parrayToMol Chemical("CCCCC")
parrayTo3D macro
parrayTo3D Chemical("CCCCC")
Torsion Scan
build smiles "C(=CC=CC1CC(=CC=CC2)C=2)C=1"
torScan v_//T [5] 5. no
show torsionProfile
convert2Dto3D macro: converting molecules from a 2D mol file or chemical spreadsheet to optimized 3D object(s)
Algorithm
convert3Dto3D macro: converting a 3D chemical from a chemical spreadsheet or 3D mol/sdf file without changing coordinates
makePharma macro
icmMacroShape: generating a smooth low resolution surface of a macromolecule.
The surface can be further colored to highlight the depth with the
occlusion shading using the color accessibility .
icmPocketFinder macro
read pdb "2hiw"
convertObject a_ yes no yes yes
icmPocketFinder a_1 4.6 yes no
show POCKETS
evalSidechainFlex macro
optimizeHbonds macro
mergePdb macro
Prev
ToxScoreHome
UpNext
Files