 Click to enlarge image.
Click to enlarge image.
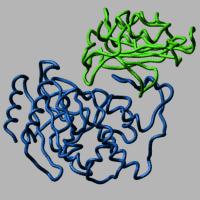
ICM-Pro contains a well validated and successful protein-protein docking algorithm. ICM-Pro also contains an algorithm to predict protein-protein docking interaction sites. In the past ICM has been used to dock ab initio a full-atom model of lysozyme to an antibody with 1.6A accuracy (Nature Struc.Biol., 1994, 1,259). Later, Maxim Totrov and Ruben Abagyan correctly predicted the association of beta-lactamase and its protein inhibitor in the Docking Challenge (Nature Struc.Biol., 1996,3,290) using the ICM pseudo-Brownian docking with subsequent ICM side-chain refinement. Read more about the docking methodology. Return to Main ICM-Pro Page
| Protein-Protein Interaction Site Prediction |
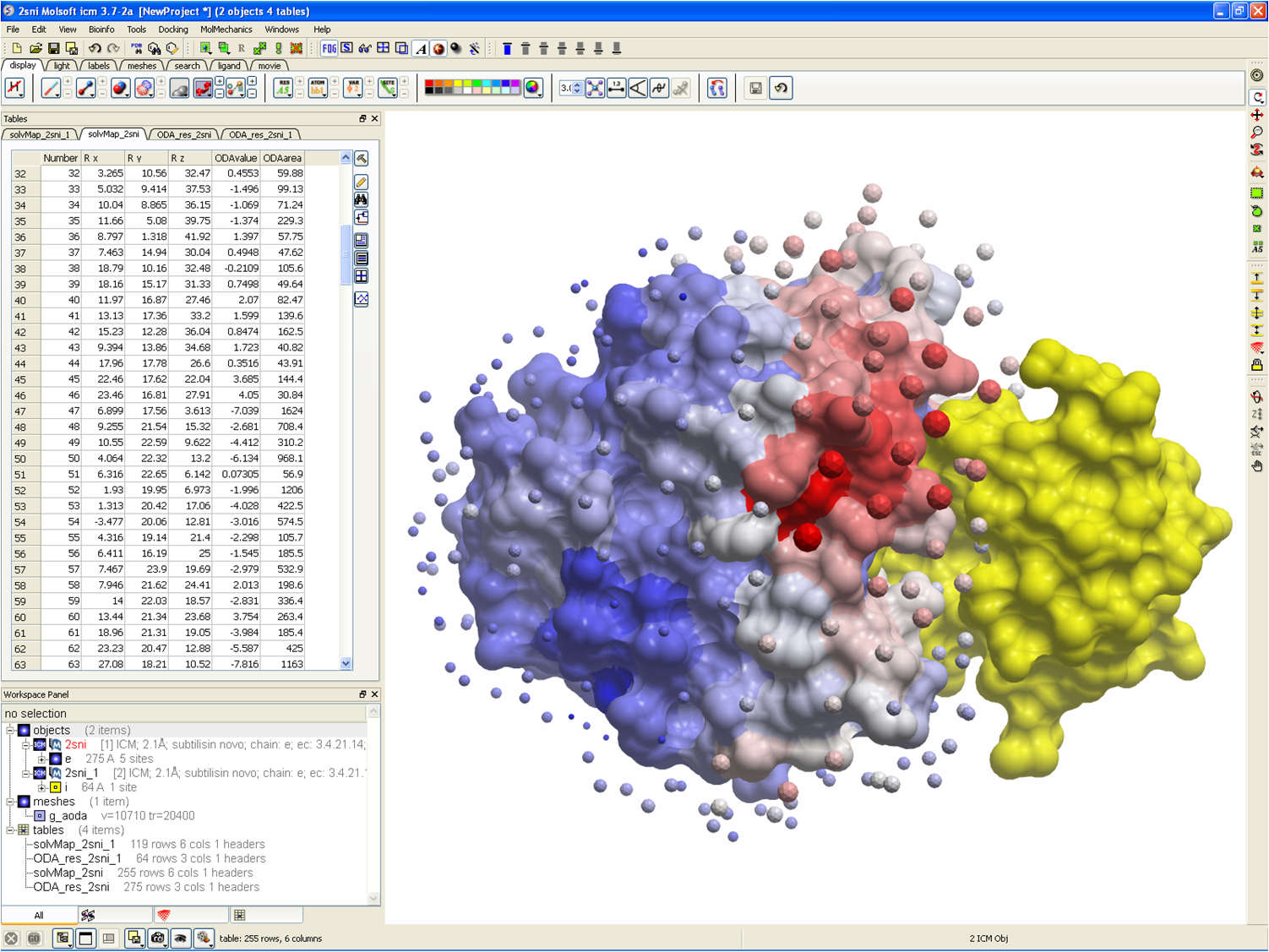
| Protein-protein interaction site prediction using the ICM Optimal Docking Area (ODA) method. |
| Color the protein by protein-protein interaction hotspots. |
| Tabulate the ODA prediction for each residue in a fully-interactive table. |
| References: Fernandez-Recio et al 2005 |
| Protein-Protein Docking |
| Dock to specific epitopes on the surface of the receptor. |
| Sample only specific epitope sites on the ligand. |
| Dock to the whole receptor (if no binding site is predicted). |
| Flexible ligand and receptor represented by maps. |
| Flexible ligand-recptor refinement. |
| Run multiple jobs on different machines in a cluster. |
| Ranked results table with energy values displayed including - van der Waals grid potential, hydrogen bonding grid potential, electrostatics grid potential, hydrophobic potential, polar terms of the solvation energy, aliphatic terms of the solvation energy, romatic terms of the solvation energy and a weighted total of the solvation energy terms. |
© 2026 All Rights Reserved MolSoft LLC Terms of Use | Privacy Policy



