GPU-Accelerated Molecular Dynamics in ICM-Pro
MolSoft
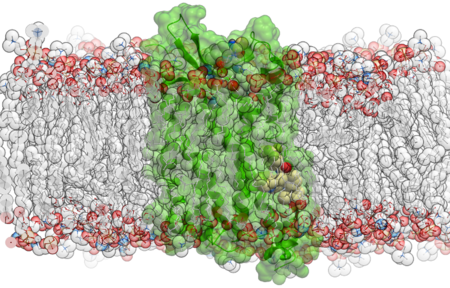
ICM-Pro includes built-in support for
OpenMM, a powerful GPU-accelerated molecular dynamics (MD) engine (
Eastman et al., 2017 ).
With optimized GPU code, ICM-Pro enables fast MD simulations—achieving 3–12 ps/s (or 0.2–1.0 μs/day) on a cost-effective workstation GPU (e.g., a standard ‘gamer’ box). ICM-Pro features a direct binary interface to OpenMM, providing:
- Seamless transfer of ICM molecular objects to the OpenMM MD engine.
- Automatic collection of MD trajectory snapshots into the ICM conformational stack for further analysis and visualization.
This tight integration delivers a user-friendly and efficient workflow for running and analyzing molecular dynamics simulations within the ICM environment.
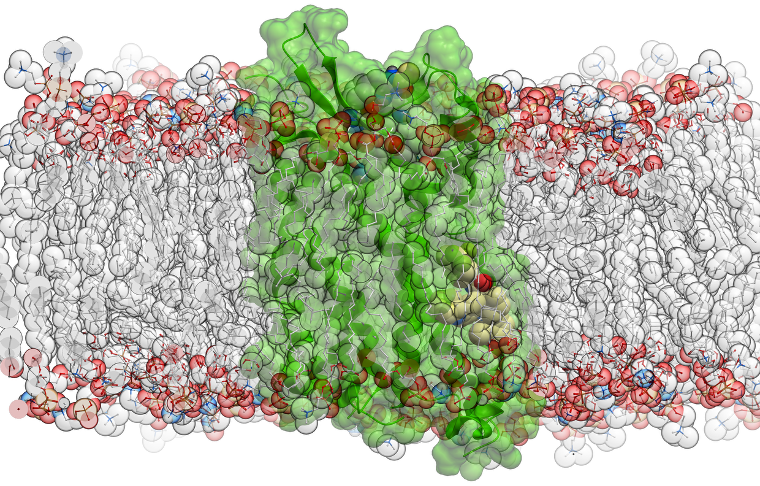
Automated MD System Preparation in ICM-Pro

MolSoft ICM-Pro streamlines molecular dynamics setup with a script and in the Graphical User Interface that encapsulates all essential system preparation steps for OpenMM simulations. The workflow includes:
- Solvating the system in a periodic water box
- Assigning AMBER atom types and charges
- Generating nnAM1/BCC partial charges for ligands
- Performing an initial energy relaxation
- Applying optional restraints as needed - tethers and distance restraints
- Using the AMBER ff14SB force field for proteins
- Support for membrane systems
This automated setup ensures consistency and efficiency, allowing you to move quickly from structure to simulation-ready system—all within the ICM environment.
Trajectory Analysis and Visualization in ICM-Pro
MolSoft
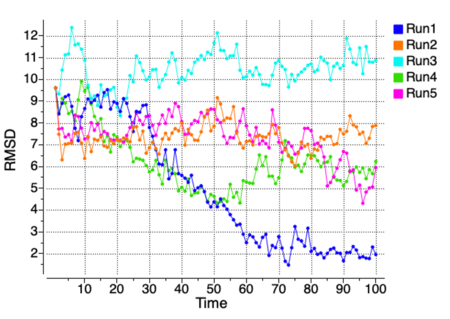
ICM-Pro provides built-in tools for analyzing and visualizing MD trajectories directly from the ICM conformational stack. A range of structural and energetic parameters can be computed and plotted over time, including:
- Interatomic distances
- RMSD to a reference structure
- RMSF (per-residue fluctuations)
- Contact area and interaction types
- Side-chain torsions (e.g., chi1 angles)
- RTCNN machine-learning score
- Physics-based scoring functions
- GB/SA binding free energy estimates (ΔGbind)
- Radius of gyration
Batch MD Simulation for VLS Hitlist Validation:
- MD relaxation of each docked complex
- Collection of multiple conformers per complex
- Tabulation of RMSD and RTCNN score drift across frames
Additional features include:
- Snapshot clustering to identify dominant conformational states
- Movie creation for visualizing dynamic behavior over time
These capabilities make ICM-Pro a complete environment for both running and interpreting molecular dynamics simulations.
 MolSoft ICM-Pro includes built-in support for OpenMM, a powerful GPU-accelerated molecular dynamics (MD) engine ( Eastman et al., 2017 ).
With optimized GPU code, ICM-Pro enables fast MD simulations—achieving 3–12 ps/s (or 0.2–1.0 μs/day) on a cost-effective workstation GPU (e.g., a standard ‘gamer’ box). ICM-Pro features a direct binary interface to OpenMM, providing:
MolSoft ICM-Pro includes built-in support for OpenMM, a powerful GPU-accelerated molecular dynamics (MD) engine ( Eastman et al., 2017 ).
With optimized GPU code, ICM-Pro enables fast MD simulations—achieving 3–12 ps/s (or 0.2–1.0 μs/day) on a cost-effective workstation GPU (e.g., a standard ‘gamer’ box). ICM-Pro features a direct binary interface to OpenMM, providing:
 MolSoft ICM-Pro streamlines molecular dynamics setup with a script and in the Graphical User Interface that encapsulates all essential system preparation steps for OpenMM simulations. The workflow includes:
MolSoft ICM-Pro streamlines molecular dynamics setup with a script and in the Graphical User Interface that encapsulates all essential system preparation steps for OpenMM simulations. The workflow includes:
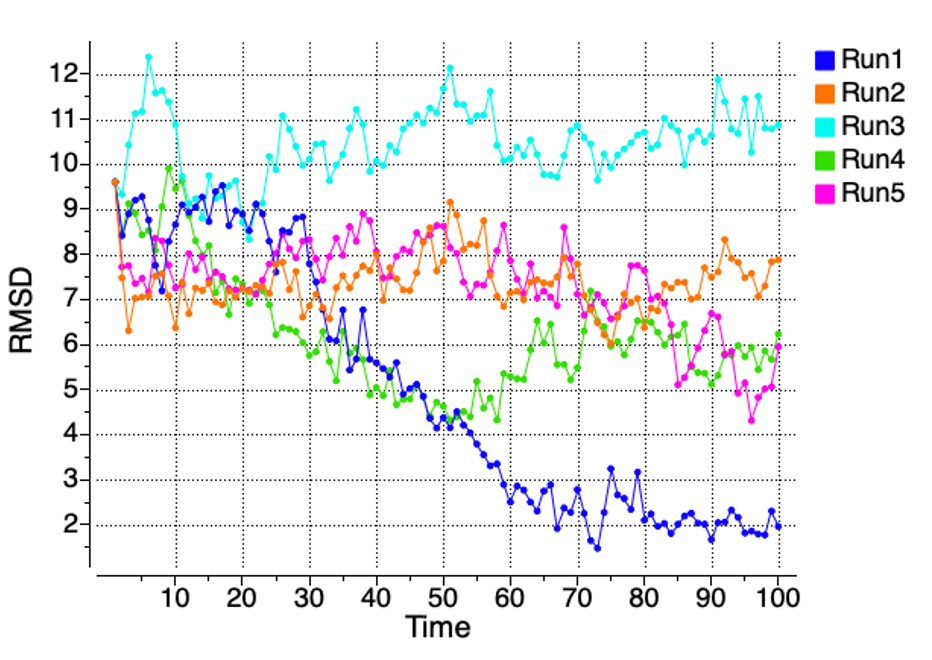 MolSoft ICM-Pro provides built-in tools for analyzing and visualizing MD trajectories directly from the ICM conformational stack. A range of structural and energetic parameters can be computed and plotted over time, including:
MolSoft ICM-Pro provides built-in tools for analyzing and visualizing MD trajectories directly from the ICM conformational stack. A range of structural and energetic parameters can be computed and plotted over time, including:



