NOTE: This product is an add-on to ICM-Pro.
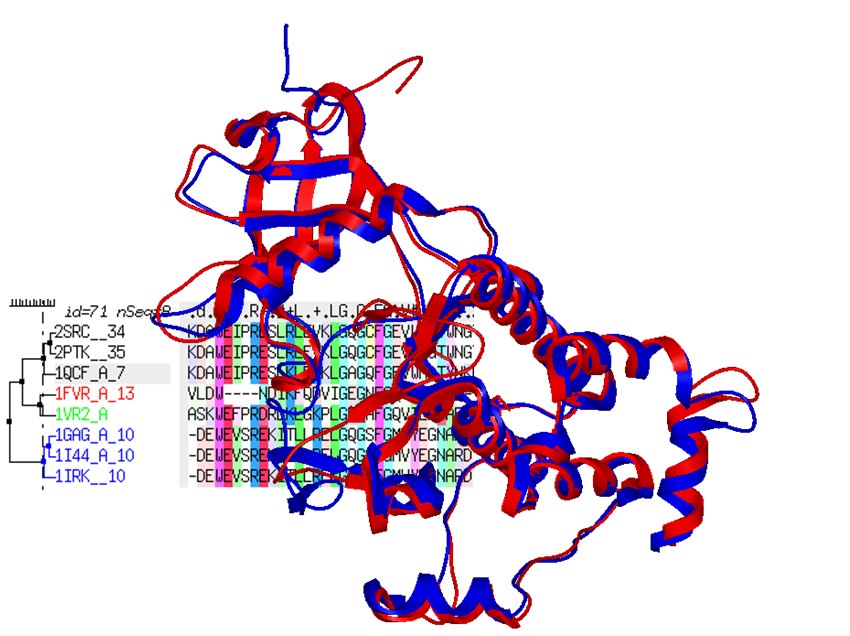
The ICM-Homology modeling algorithm has proved to be one of the more robust modeling tools (1-6). After initial placement of the aligned polypeptide chain onto the template structure, the side-chain torsion angles are predicted by simultaneous global optimization of the energy for all non-identical residues.
Methodology for conformational modeling of protein side chains and loops, implemented in ICM, relies on internal coordinate definition of the molecular object combined with computationally efficient ICM Biased Probability Monte Carlo (BPMC) optimization (7). An extended force field includes surface terms, electrostatics with the boundary element solution of the Poisson equation (8), side chain entropy terms, and a fast algorithm for calculating molecular surfaces. The quality of the resulting 3D model is assessed by a specialized ICM procedure, which also predicts possible backbone deviations between the homologues.
The ICM homology modeling algorithm has demonstrated excellent accuracy in blind predictions at the CASP2 competition 6 and in several protein engineering applications (9-16). Moreover, recent results show that ICM models built with as little as 35% identity can be accurate enough to be successfully used in receptor based rational drug design (1-6,17).
Key features of ICM-Homology include:
References
(1) Schapira, M., B.M. Raaka, H.H. Samuels, andR. Abagyan. 2001. In Silico discovery of novel Retinoic Acid Receptor agonist structures. BMC Structural Biology 1:1.
(2) Schapira, M., R. Abagyan, andM. Totrov. 2002. Structural model of nicotinic acetylcholine receptor isotypes bound to acetylcholine and nicotine. BMC Struct Biol 2(1):1.
(3) Schapira, M., R. Abagyan, andM. Totrov. 2003. Nuclear hormone receptor targeted virtual screening. J Med Chem 46(14):3045-3059.
(4) Schapira, M., B.M. Raaka, S. Das, L. Fan, M. Totrov, Z. Zhou, S.R. Wilson, R. Abagyan, andH.H. Samuels. 2003. Discovery of diverse thyroid hormone receptor antagonists by high-throughput docking. Proc Natl Acad Sci U S A 100(12):7354-7359.
(5) Schapira, M., B.M. Raaka, H.H. Samuels, andR. Abagyan. 2000. Rational discovery of novel nuclear hormone receptor antagonists. Proc. Natl. Acad. Sci. U. S. A. 97(3):1008-1013.
(6) Abagyan, R., S. Batalov, T. Cardozo, M. Totrov, J. Webber, andY.Y. Zhou. 1997. Homology modeling with internal coordinate mechanics: Deformation zone mapping and improvements of models via conformational search. Proteins:29-37.
(7) Abagyan, R.A., and M.M. Totrov. 1996. Biased probability Monte Carlo as a powerful global energy optimization method for biomolecular structure prediction. Abstr. Pap. Am. Chem. Soc. 211:35-COMP.
(8) Totrov, M., andR. Abagyan. 2001. Rapid boundary element solvation electrostatics calculations in folding simulations: successful folding of a 23-residue peptide. Biopolymers 60(2):124-133.
(9) Borchert, T.V., R. Abagyan, R. Jaenicke, andR.K. Wierenga. 1994. Design, Creation, and Characterization of a Stable, Monomeric Triosephosphate Isomerase. Proc. Natl. Acad. Sci. U. S. A. 91(4):1515-1518.
(10) Borchert, T.V., K.V. Kishan, J.P. Zeelen, W. Schliebs, N. Thanki, R. Abagyan, R. Jaenicke, andR.K. Wierenga. 1995. Three new crystal structures of point mutation variants of monoTIM: conformational flexibility of loop-1, loop-4 and loop-8. Structure 3(7):669-679.
(11) Norledge, B.V., A.M. Lambeir, R.A. Abagyan, A. Rottmann, A.M. Fernandez, V.V. Filimonov, M.G. Peter, andR.K. Wierenga. 2001. Modeling, mutagenesis, and structural studies on the fully conserved phosphate-binding loop (loop 8) of triosephosphate isomerase: Toward a new substrate specificity. Proteins 42(3):383-389.
(12) Thanki, N., J.P. Zeelen, M. Mathieu, R. Jaenicke, R.A. Abagyan, R.K. Wierenga, andW. Schliebs. 1997. Protein engineering with monomeric triosephosphate isomerase (monoTIM): the modelling and structure verification of a seven-residue loop. Protein Eng 10(2):159-167
(13) Cardozo, T., Totrov, M.M. & Abagyan, R.A (1995) Homology modeling by the ICM method. Proteins, 23, 403-414.
(14) Rashin, A.A., Rashin, B.H., Rashin A., Abagyan, R. (1997) Evaluating the energetics of empty cavities and internal mutations in proteins. Protein Science, 6, 2143-2158.
(15) Norledge, B.V., Lambeir, A.M., Abagyan, R.A., Rottmann, A., Fernandez, A.M., Filimonov, V., Peter, M.G., and Wierenga, R.K. (2001). Modeling, mutagenesis, and structural studies on the fully conserved phosphateloop (loop 8) of triosephosphate isomerase: toward a new substrate specificity. Proteins Feb 15;42(3), 383-9
(16) Cardozo, T.J., Abagyan, R. (1998) Molecular Modeling of the Domain Shared Between CED-4 and its Mammalian Homologue Apaf-1: A Structural Relationship to the G-proteins. J. Mol. Model., 4, 83-93.
(17) Abagyan, R., andM. Totrov. 2001. High-throughput docking for lead generation. Curr Opin Chem Biol 5(4):375-382.
© 2026 All Rights Reserved MolSoft LLC Terms of Use | Privacy Policy



