| Prev | ICM User's Guide 4.4 Protein Superposition | Next |
[ Display and Select Proteins for Superposition | Superimpose Button | Proteins by 3D | Multiple Proteins | Arrange as Grid | Superimpose by APF ]
| Available in the following product(s): ICM-Browser-Pro | ICM-Pro |
 One or more proteins can be superimposed. Simply select the molecules or parts of the molecules you wish to superimpose and then use the selection of
protein superimpose tools described in this section. A convenient superimpose button can be found in the Display tab (see image of button (left).
One or more proteins can be superimposed. Simply select the molecules or parts of the molecules you wish to superimpose and then use the selection of
protein superimpose tools described in this section. A convenient superimpose button can be found in the Display tab (see image of button (left).
Chapter Contents:
- Select Proteins for Superposition
- Superimpose Button
- Superimpose by 3D
- Superimpose Multiple Proteins
- Arrange as Grid
- Superimpose Sites by Atomic Property Fields
4.4.1 Select Proteins for Superposition |
Before any superposition operation can be undertaken you need to select the protein structures you wish to superimpose.
One way to do this is by selecting in the ICM workspace. For other selection tools please see the Making Selections section of the manual.
- Select both receptors by double clicking on the name of the molecule in the ICM Workspace. To select two molecules use the Ctrl button or use the shift button to select a range of objects in the ICM Workspace. A receptor which is selected will be highlighted in blue in the ICM Workspace and with green crosses in the graphical display.
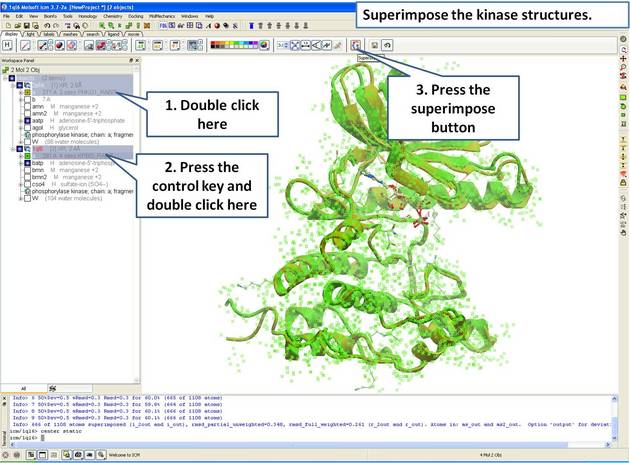
Once the molecules are selected you can then superimpose them using the options described in the next section of this manual.
4.4.2 Superimpose Button |
A convenient way to superimpose two molecules is by using the superimpose button in the display tab, ICM will calculate the Ca-atom, backbone atom and heavy atom differences between the two structures. More advanced superimpose options can be found in the Tools/Superimpose menu.
To superimpose:
- First load the two structures into ICM.
- Select which parts or all of the two structure you wish to superimpose (see the chapter on Selections or the description protein-superposition-select{here}.).
- Select the display tab (previously called Advanced tab) at the top of the GUI.
- Select the superimpose button.
The rmsd will be displayed in the terminal window as shown below:

| NOTE: You do not need to select the whole molecule, the superimpose button will work on small selections e.g the loop regions or domains. |
4.4.3 Superimpose by 3D |
To superimpose proteins by 3D:
- First display and select the proteins you wish to superimpose by 3D.
- Tools/Superimpose/Proteins by 3D
- A window as shown below will be displayed.
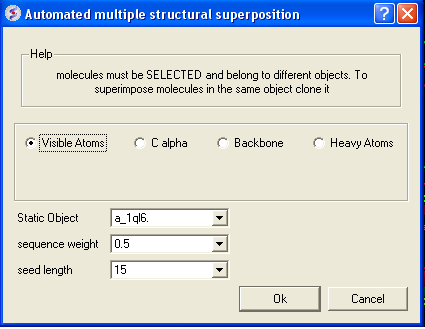
- Select by which atoms you wish to superimpose.
- Enter the ICM selection language description for the protein structure you wish to remain static. You can also use the drop down arrow button to select it.
- Enter the sequence weight Average local sequence alignment score.
- Enter the seed length This is the similarity window size.
4.4.4 Superimpose Multiple Proteins |
To superimpoe multiple proteins:
- First display and select the proteins you wish to superimpose by 3D.
- Tools/Superimpose/Multiple Proteins
- A window as shown below will be displayed.
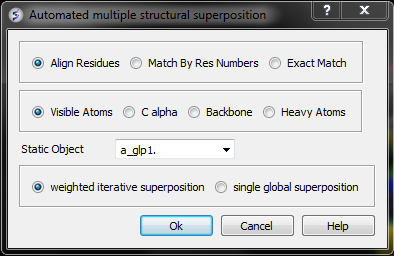
- Select by which method you would like to superimpose
Align Residues - Residue correspondence is established by sequence alignment using the ICM ZEGA alignment Abagyan, Batalov, 1997. Atom alignment: by atom name.
Match by Res Numbers - Residue alignment by residue number.Atom alignment: by atom name for pairs of identical residues or pairs of close residues (F with Y; B with D,N; D with N; E with Q or Z, Q with Z), for other residue pairs only the backbone atoms ca,c,n,o,hn,ha are aligned.
Exact Match - Residue alignment is by the Needleman and Wunsch method. Inside residue atoms are aligned sequentially and regardless of the name.
- Select which atoms you would like to superimpose. Visible Atoms, C alpha, Backbone, or Heavy Atoms.
- Select whether you would like to use weighted iterative superposition as described here http://www.molsoft.com/man/icm-commands.html#superimpose-minimize or non-iterative single global superposition.
4.4.5 Arrange as Grid |
To separate superimposed proteins:
- Tools/Superimpose/Arrange as Grid

4.4.6 Superimpose Sites by Atomic Property Fields |
Here we describe how to superimpose and compare a ligand binding site using Atomic Property Fields. This method is described in more detail in this publication.
In this example we superimpose the ligand binding pocket of thiamine diphosphate in the binding sites of pyruvate dehydrogenase (pdb code: 1rp7) and pyruvate decarboxylase (pdb code:1pvd).Even though the sequence identity between both proteins is very low (19%) and the secondary structure surrounding the ligand undergoes considerable displacement you will see that the pockets can still be superimposed very well using the APF method.
Some Comments about Selecting the Pocket APF pocket overlay works by superimposing a layer of receptor atoms around the pocket using APF chemical property fields of these atoms "pocket" of course is something that is not uniquely defined and is somewhat subjective. One way to define it is to use the ligand inside, in which case a layer of receptor atoms within a certain cutoff distance from the ligand is considered in superposition. This is "around selected ligand" mode. It focuses on atoms that directly interact with the ligand while largely disregarding backbone fold, which can be helpful when comparing different pockets binding similar ligands. The other mode uses input selections directly, leaving it up to the user to define the sets of atoms that comprise the two pockets. Depending on the system and degree of similarity, different selections may work better or worse. For instance in the absence of ligand one may want to detect pocket with pocket finder and then select residues around the pocket "blob". Regarding the influence of ligand on superposition, if pocket is defined via the ligand it will of course matter whether the ligands extend into certain regions or not. If external pocket definition is used, ligands will have no direct effect on superposition process.
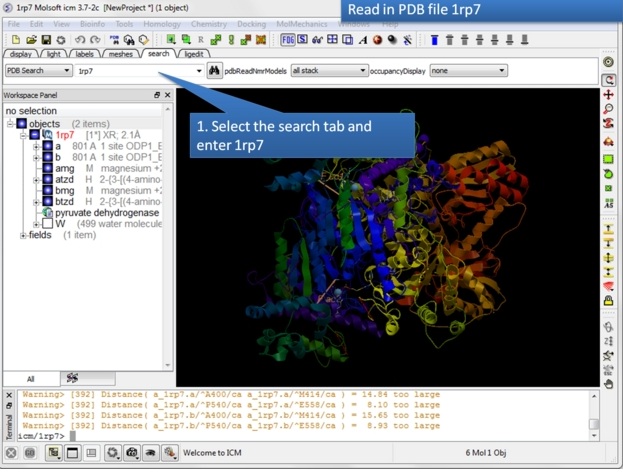 |
| Step 1: Read in the pyruvate dehydrogenase (pdb code: 1rp7) structure. |
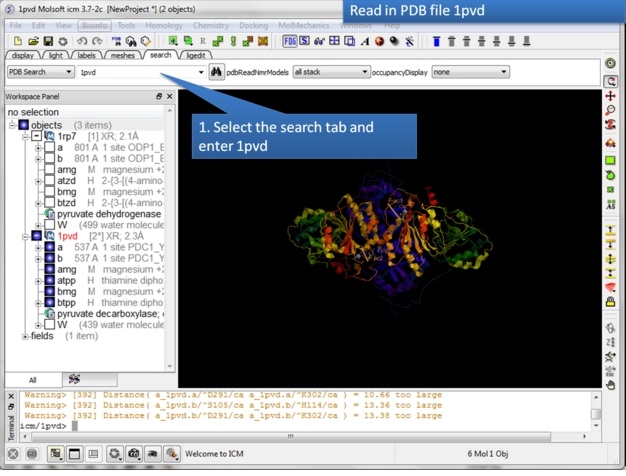 |
| Step 2: Read in the pyruvate decarboxylase (pdb code:1pvd). |
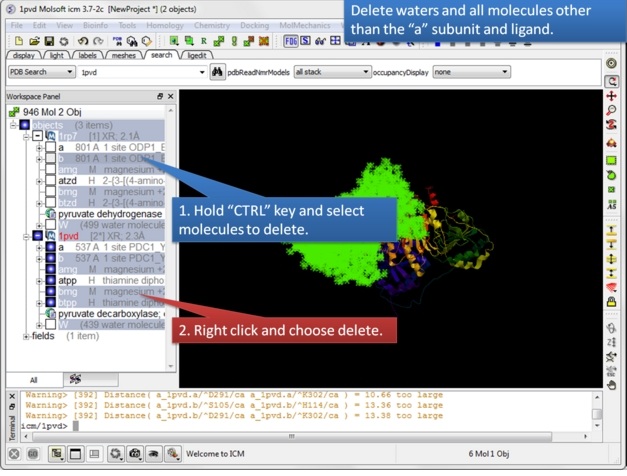 |
| Step 3: Delete unwanted molecules in the b chain and waters. |
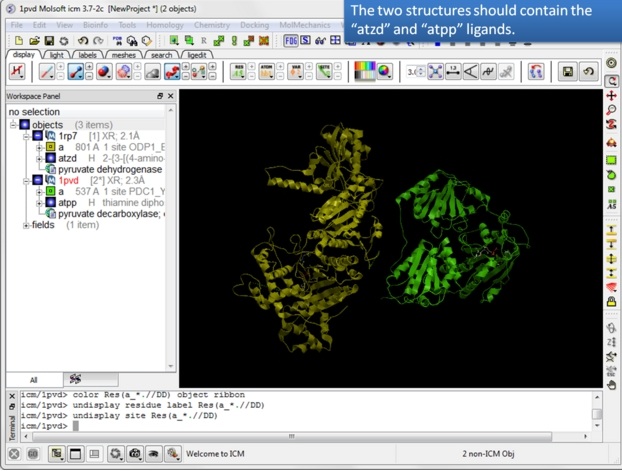 |
| Step 4: You should have two objects containing the protein and ligand. |
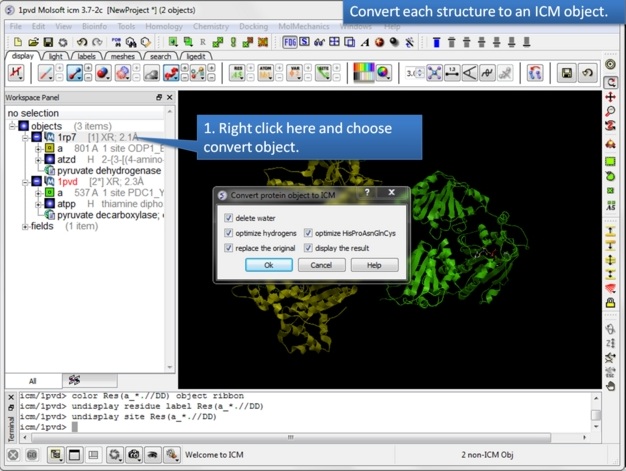 |
| Step 5: Convert both structures to an ICM object. |
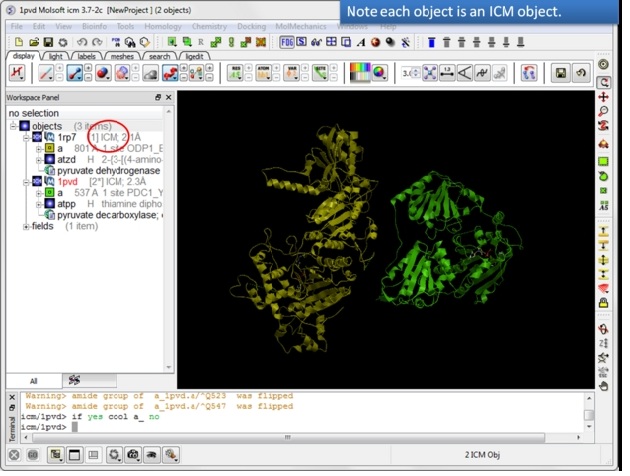 |
| Step 6: If the pdb is converted you will see "ICM" in the workspace. |
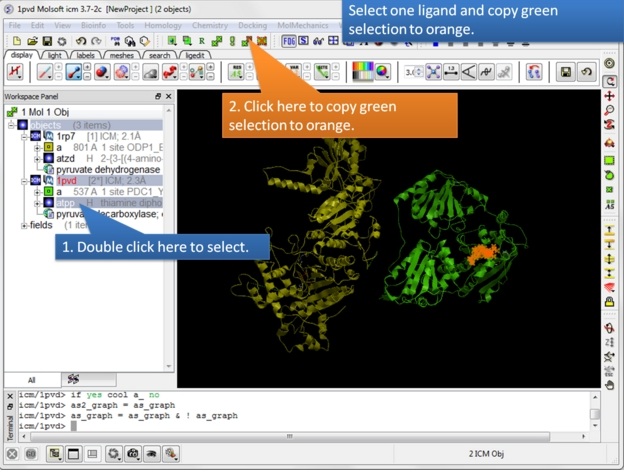 |
| Step 7: Select one ligand and copy the green selection to orange. |
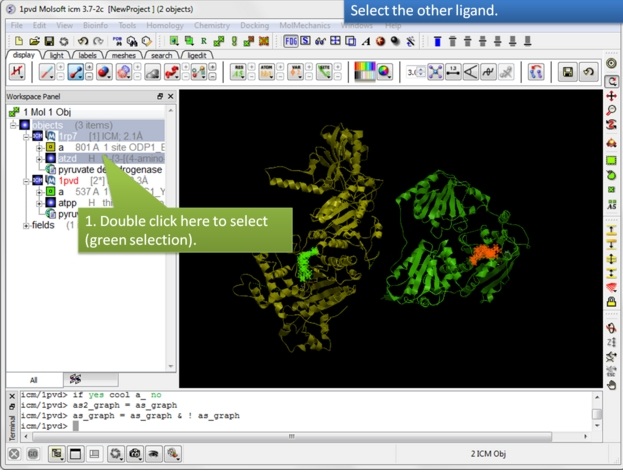 |
| Step 8: Select the other ligand (regular green selection). |
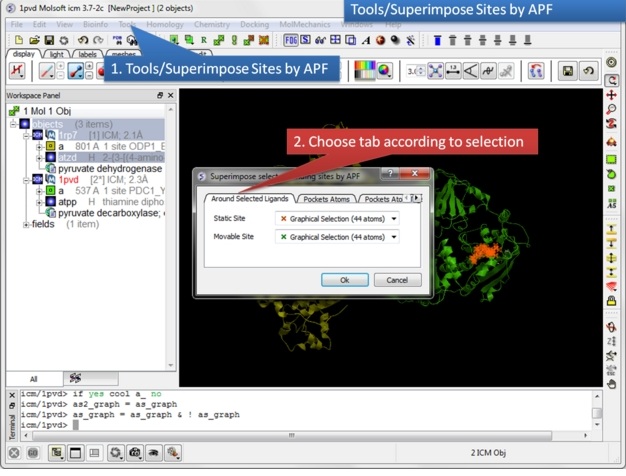 |
| Step 9: Select the option Tools/Superimpose/Sites by APF. Note you can also superimpose pockets - choose the appropriate tabs in the dialog box. |
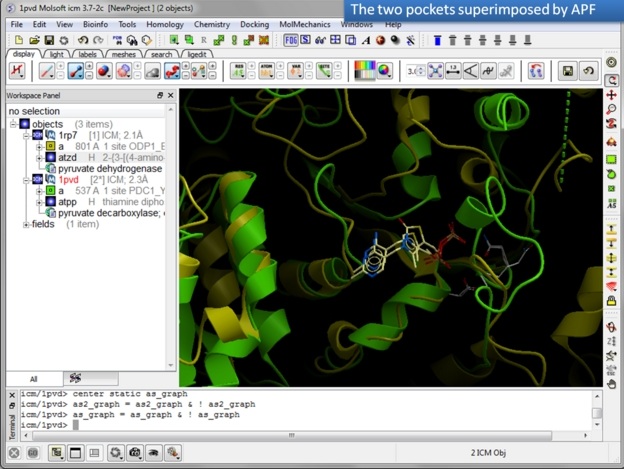 |
| Step 10: Observe the superimposed site. |
| Prev Convert Xray Density to Grid | Home Up | Next Protein Structure Analysis |