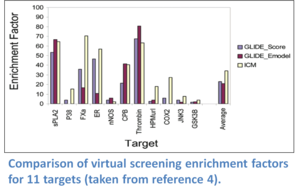
ICM virtual ligand screening technology has been ranked the best virtual screening tool in comparisons reported by the Scripps Research Institute (Bursulaya et al 2003), Astra Zeneca (Chen et al 2003), and Wyeth (Cross et al 2009). ICM-VLS ranked number one in terms of predicting the ligand pose and enrichment factor (number of compounds you need to test experimentally to find a hit) compared to a selection of other commercially available screening algorithms. For example, scientists at Astra Zeneca screened a database containing 20K random compounds and between 17-622 active ligands per drug target receptor. The enrichment factors for 12 targets at 1% of database subset compared to Schrodinger's Glide software is shown below. ICM molecular modeling and docking also performed very well at "blind" GPCR modeling and docking competitions (See (Michino et al 2009), (Katritch et al 2010), (Kufareva et al 2010) ).
MolSoft's docking and scoring algorithm ranked first place for prediction of ligand pose and screening accuracy in the most recent industry-wide competition organized by OpenEye, GlaxoWellcome, and Merck. The results of the competition were announced at the American Chemical Society Meeting in Anaheim in 2011 and MolSoft's ICM performance is reported here (Neves et al 2012). The docking pose prediction accuracy was benchmarked using the modified Astex set of 85 protein-ligand complexes. The top score poses were correct (under 2A RMSD) in 60% to over 90% of the cases depending on the docking method. The ICM docking method achieved 78% of the top score poses under 1A RMSD and 91% under 2≈ RMSD.
MolSoft's ICM software ranked in first place for average RMSD docking accuracy in the 'Blind - Industry Wide' Drug Design challenge competition http://drugdesigndata.org/ (D3R).
Along with over 50 other participants, the Molsoft group led by Maxim Totrov Ph.D.(Principal Scientist, MolSoft), submitted blind docking pose predictions for the Farnesoid X receptor (FXR) which is a drug target for dyslipidemia and diabetes. The MolSoft team used the pocketome (www.pocketome.org) entry for FXR and the ICM-VLS, Atomic Property Fields (APF) and machine learning methods in the ICM-Pro software to predict the interaction of 36 FXR ligands and the binding affinity of 102 other ligands.
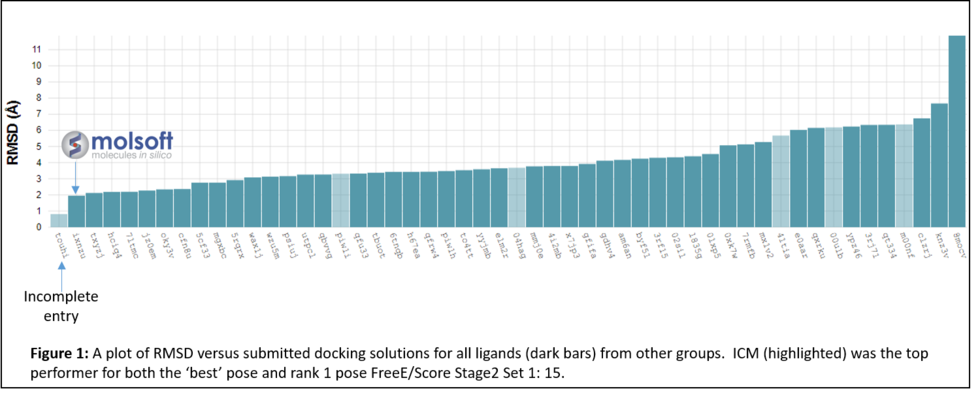
In January 2017 the organizers (D3R) distributed the evaluation results which showed that MolSoft's submission ranked first in RMSD for the top scoring pose and was the only one with average RMSD below 2.0A (Figure 1 below). In the Binding Energy Prediction competition ICM APF dock and icm-MMGBSA method ranked in first place and produced the lowest RMSE for Stage 2 Set 1 set of Kd values for 15 ligands (Lam et al 2018).
In the 2018 Grand Challenge 3 there were Six different targets Cathepsin S and kinases VEGFR2, JAK2 and p38-alpha. MolSoft's submissions ranked first place for docking pose and affinity prediction for all targets Lam et al 2019. In the 2019 Grand Challenge 4 ICM accurately predicted the pose of BACE macrocycle inhibitors to within sub-angstrom accuracy Lam et al 2019.
© 2026 All Rights Reserved MolSoft LLC Terms of Use | Privacy Policy



