[ Energy Units | Calculate Binding Energy | Faq guided dock | Faq reload dock | Faq dock repeat | SCORE | Faq hitlist | Faq score | Ligand and box | Pocket Finder | Dock Time | Thoroughness | Probe | Faq receptor selection | Induced Fit | Dock Command Line | Faq scanScoreExternal | Faq scanScoreExternal2 | Background Job | flexible rings | Dock Racemic | Dock charge groups | Working directory | Retrieve columns ]
Frequently asked questions regarding small molecule and protein-protein docking.
- What are the units of the energy values displayed after docking?
- I do not have ICM-VLS but I would like to calculate the binding energy of my docked complex - how can I do this?
- How can I guide my docking to a known conformation of a smilar ligand?
- How do I reload a docking project?
- During a Virtual Ligand Screening experiment how many times should I re-run the docking?
- Which score value should I use for analysis?
- Some compounds are missing from my HITLIST.
- What constitutes a good docking score?
- When I view my docking run my ligand never jumps into the box - what did I do wrong?
- How do I identify the binding pockets in my receptor?
- How long does it take to dock one ligand using ICM-VLS?
- What does thoroughness or effort mean?
- When I setup the receptor I am asked to move a probe - what is this?
- I want to dock to the receptor and include other molecules in the receptor such as a tightly bound water molecule - how can I do this?
- How can I run docking with a flexible receptor?
- How can I run the docking simulation from the UNIX command line?
- I have a complex I wish to generate an ICM VLS Score for, however I did not dock it using VLS. How can I do this?
- Why is there always a small difference between the score calculated interactively by scanScoreExternal and that obtained by docking (VLS)?
- How do I monitor and terminate a background docking job?
- How do I sample flexible ring conformations (boat, chair etc..) during docking?
- I am docking a racemic compound how can I sample both R and S states during docking?
- What do the options mean in Docking/Preferences/Charge Groups?
- How do I change the working directory?
- After VS how do I retrieve columns from my original database and add them to the hitlist?
The energy units are kcal/mol. The value reported in the SCORE is unitless.
For more in depth information on this topic please see the command line manual www.molsoft.com/man however the basic approach is this: Calculuate the energy of the receptor (e1, a_1), energy of the ligand (e2, a_2) and the energy of the complex (e12) then the binding energy = e12 - e1 - e2
Here is a script to do this whereby the ligand is a_2 and the receptor is a_1:
electroMethod="boundary element"
surfaceMethod="constant tension"
surfaceTension=0.020
dielConst = 12.7
set terms "sf,el,en"
read object s_icmhome+"2ptc"
show energy a_1 a_1 mute
e1 =Energy("el,sf,en")
show energy a_2 a_2 mute
e2 =Energy("el,sf,en")
show energy mute
e12 =Energy("el,sf,en")
print "Binding energy = ", e12 - e1 - e2There are many different approaches to the evaluation of binding energy. One of the reasonable approximations has the following features: van der Waals/hydrogen bonding interaction is excluded since it has close magnitudes for protein-protein and for protein-solvent interactions; electrostatic free energy change is calculated by the REBEL method (see also the section "How to calculate the electrostatic free energy ... ") above); side-chain entropy change is calculated by standard ICM entropic term based on exposed surface area of flexible side-chains; hydrophobic energy change is calculated using surface term with constant surface tension of 20. cal/Angstrom.
Use Docking/Template as described here.
Please see this section.
Generally we suggest the docking should be repeated 2-3 times and the lowest ICM score pose should be taken.
The value under the heading SCORE relates to the ICM docking score and is the best one to use for docking result analysis. The other score we provide - potential of mean force score (mfscore - http://www.molsoft.com/man/terms.html#term-mf) provides an independent score of the strength of ligand-receptor interaction.
The hitlist is filtered according to a score cutoff defined in the PROJECTNAME.tab or .dtb file. Therefore poor scoring compounds are not reported in the HITLIST - this can be changed by opening the .dtb file in a suitable text editor e.g. notepad in windows or vi and changing the DOCK1.r_ScoreThreshold value (by default it is -32). The scores for all compounds in a VLS screen are available in the PROJECTNAME.OU file.
You can also change this value in Graphical User Interface:
- Docking/Preference/Database Scan
- Change the Score Threshold Value
Generally a score below -32 is regarded as a good docking score. A good score depends on the system into which you are docking. Buried hydrophobic pockets will likely return low scores but for others a score higher than -32 might be reasonable. If a co-crystal structure is available remove the ligand and re-dock it to get an indication of approximately what is a good score for the receptor you are interested in.
Here are some reasons and some solutions for why your ligand is sampling outside of the binding pocket:
- On Receptor Setup when it asks to move the initial probe - did you accidentally move the probe outside the box?
- Double check exactly where you built the maps (read one map in) in command line type read map "DOCK1_gl" ds map or check Docking/Review Adjust Ligand binding box
- On the Docking/Interactive Docking/ LoadedLigand - did you check the box Use Current Ligand position? If so remove this option.
To do this go to:
- Tools/3D Predict/ICMPocketFinder
More information can be found in the section entitled Identifying Binding Pockets
It takes approximately 10-30 seconds per ligand depending on the size of the ligand and the nature of the pocket.
When you send a docking job either using Docking/Interactive or Docking/Run Docking Batch you are asked to enter a thoroughness value (was changed to effort after 3.8-2 release). This value represents
the length of the docking simulation. The default value is 1. and this works well with nearly every kind of docking scenario, however in certain circumstances such as if you have a very large pocket this value should be increased slightly to a range of between 5. and 10.
The probe which you see after Receptor Setup (Docking/ Receptor Setup) represents the initial starting position for the ligand. Usually this does not have to be changed as ICM by default places it into the center of the pocket. However if you do wish to move it to position closer to a critical region of the pocket you can do this using the middle mouse button when prompted in receptor setup.
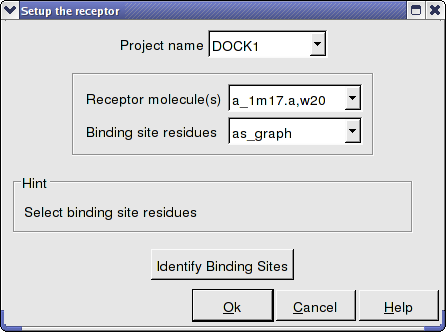
All molecules that you wish to dock to need to be stated in the Docking/Receptor Setup/ Receptor molecule (s) data entry box using the ICM selection language. For example if you wish to dock to the protein with PDB code 1m17 and water molecule number 20 you need to enter the information as shown below:
There are several options available for induced fit docking and they are described here
Use dockScan as described here: http://www.molsoft.com/man/ligand-fit.html#dockScan
type in the command line:
scanScoreExternalOR
- Docking/Tools/Evaluate ICM Score...
The reason for this is that ICM score has terms that require calculations on the reference 'free' state of the ligand, in particular solvation electrostatics and internal force-field strain energy change are calculated as a difference between free and bound state. VLS uses as a free state the lowest energy conformation found by MC search for the unbound ligand. Interactive score just minimizes the ligand locally. To ensure consistency we recommend you use one method or the other for scoring or you could recalculate the interactive score for your ligand from VLS before modifying/minimizing.
If a background job is running you will see a message saying "1 bgrnd job" at the top of the gui interface (gui blue title panel).
To monitor the progress of the job:
- Windows/Background Jobs
- A panel will be displayed with information such as running time and percentage completed.
To terminate a background job:
- Windows/Background Jobs
- Right click on the job ID number and select "Terminate".
To view the current output of a background job:
- Windows/Background Jobs
- Right click on the job ID number and select "View Output"
When a background job has finished a message will appear in the graphical user interface
MolSoft's ICM docking algorithm has flexible ring sampling included on the fly. Just set ring sampling level to 1 (flex ring only in pre-sampling step) or 2 (throughout the simulation).
To do this:
- Set up the docking project (http://www.molsoft.com/gui/start- dock.html#docking-start)
- Before running the docking simulation go to Docking/Preferences/ General and change the flexible ring sampling level to 1 or 2.
- Now run the docking simulation (http://www.molsoft.com/gui/start- dock.html#begin-docking-simulation)
OR,
If you want to generate the conformations before docking and you have ICM-Pro + ICM-Chemistry then you can use the conformation generator algorithm described here: http://www.molsoft.com/gui/conf-gen.html
You can read more details about the fields in the docking project .dtb or tab file here: https://molsoft.com/addons/ftp/man/dockTable.pdf
To sample both R and S states of a compound during docking. Edit the project_name.dtb file and edit l_sampleRacemin to yes
If l_sampleRacemic is 'yes', R and S states are sampled for racemic centers and best-fitting one is chosen. If it is 'no', they are kept fixed (in an aribtrary R or S state). Note that stereo centers that centers with pre-assigned R or S state are never sampled, if sampling is desired they need to be reassigned as racemic.
This relates only to the ligands:
- NH2 = ICM will charge primary amines only
- NH2 NH ICM will charge primary & secondary amines only
- NH2 NH NT ICM will charge primary & secondary & tertiary amines only
- NH2 NH NT imidazole ICM will charge primary/sec/tert and imidazoles.
In the very latest version of ICM (3.7-2a) we have a prediction of pKA which will automatically charge each ligand according to the pKA prediction. If you want to change the charge of the residues on the receptor please right click on the ligand select mutate and their are uncharged versions of each residue. If you do this then you would have to re-make the maps.
This is a common question from people who use Windows especially for docking. By default in Windows all the docking files are stored in a directory called "My Document". This may not be convenient and so the way to change this is to use the command line and type: set directory "c:/path_to_directory/"' In the very latest version of the software there is now a GUI option to do this in the Tools menu.
When I make a hitlist after VS (Docking/Make Hitlist) not all the data will be transferred from the original database. How can I retrieve this? You can do this by reading in the index of the original sdf file(e.g index = db.inx and hitlist table = hitlist_answers)
read index "//arra/andy/db/db.inx"
read table index db[hitlist_answers.IX]