[ Refinement | 4D Docking | Explicit Group Docking | SCARE ]
| Available in the following product(s): ICM-Pro | ICM-VLS | |
ICM contains a selection of tools to account for pocket flexibility (induced fit) in docking and virtual screening. You can read more about the methods here: http://molsoft.com/induced-fit.html
Currently there are four ways to incorporate induced fit into docking:
- Ligand and receptor complex refinement. This approach allows the side-chains of the receptor to be fully flexible.
- Multiple receptor conformation docking. Multiple receptor conformations can be obtained from modeling alternative conformations or from an ensemble of crystal structures.
- Explicit group docking allows certain defined residues to be flexible during the docking procedure. For example the hydroxyls of Ser, Thr, and Tyr can be treated explicitly during docking and the rest of the atoms will be treated as maps.
- The SCARE method allows you to take into account induced fit of the receptor upon ligand docking. The method systematically scans pairs of neighboring side-chains and replaces them with Alanine and docks a ligand to each of the "gapped" models.
To undertake flexible ligand and receptor refinement docking you first need to dock the ligand into the receptor using the procedures described in the previous section entitled Small Molecule Docking. The next step is to select the pose from your docking experiment you wish to use for flexible docking.
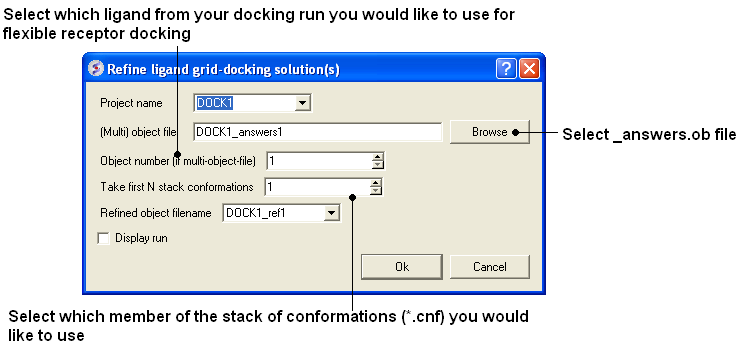
- Docking/Flexible-Receptor/Refinement and a window as shown below will be displayed.
- Enter the name of the initial docking project.
- Select the answers.ob file from the docking experiment.
- If you docked more than one ligands you need to select which ligand you want to use for the flexible receptor docking. For example if you want to refine the 3rd ligand in your docked database you would enter 3 in the Object number data entry box.
- Select which member of the stack of docking solutions you wish to use. In most cases it will be the first member of the stack and therefore you will enter "1".
- Enter a name for the refined complex.
- You can display the run in the graphical display but this will slow the process down.
To view the results: The refinement procedure creates two new files in your working directory (see Tools/ Change Working Directory for location). The files are the object with docking_project_name_ref1.ob and a stack of conformations called docking_project_name_ref1.cnf.
- File/Open docking_project_name_ref1.ob
- File/Open docking_project_name_ref1.cnf
- Select MolMechanics/Stack View and a table of conformations will be displayed.
- Click on each row to load the conformation and view it.
| Additional Resources: Tutorial: 4D Docking to Multiple Conformations of a Flexible Loop in the Aldose Reductase Ligand Binding Pocket |
Standard molecular docking treats the target protein as a rigid structure, which can lead to false negatives if a ligand requires the protein to adapt its shape (induced fit). 4D Docking (Multiple Receptor Conformation Docking) overcomes this by introducing the fourth dimension—conformational flexibility—allowing you to dock ligands into a "stack" (ensemble) of multiple receptor structures simultaneously.
To perform 4D docking, you must first generate this conformational stack. Your starting point typically falls into one of two scenarios.
- Ideal Scenario - you have an ensemble of crystal structures of the same protein.
- You have a single crystal structure and you wish to generate an ensemble of possible conformations using Monte Carlo Simulation or Molecular Dynamics of a particular region of the binding pocket. See the Aldose Reductase example which contains a flexible loop in the ligand binding pocket.
How to generate a stack of conformations for a single crystal structure: One way to do this is to select a number of side-chains and right click on the graphical selection and choose Advanced/Optimize Side Chains or right click on the ligand in the binding pocket and select Advanced/Optimize Ligand Vicinity. Or you can model the loop regions using MolMechanics/Sample Loop.
How to generate a stack from an ensemble of crystal structures for Multiple Receptor Docking: The steps to undertake multiple receptor docking (4D docking) are described below. Before you can start multiple receptor docking all your structures need to be in a stack.
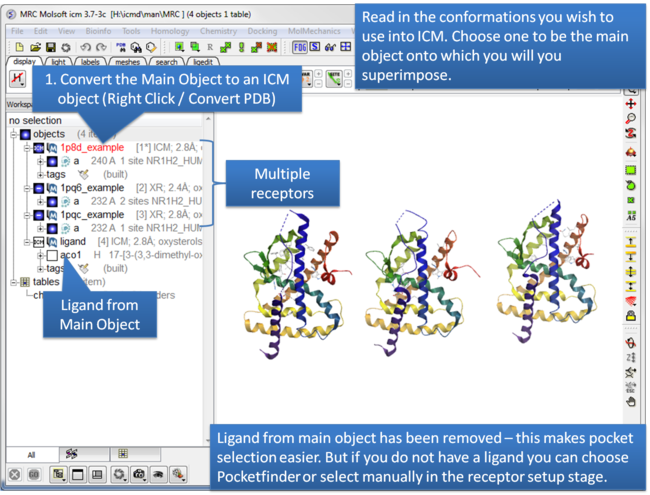 |
| Step 1: Read in the conformations of the receptor you would like to dock to. Convert the Main Object to an ICM Object. |
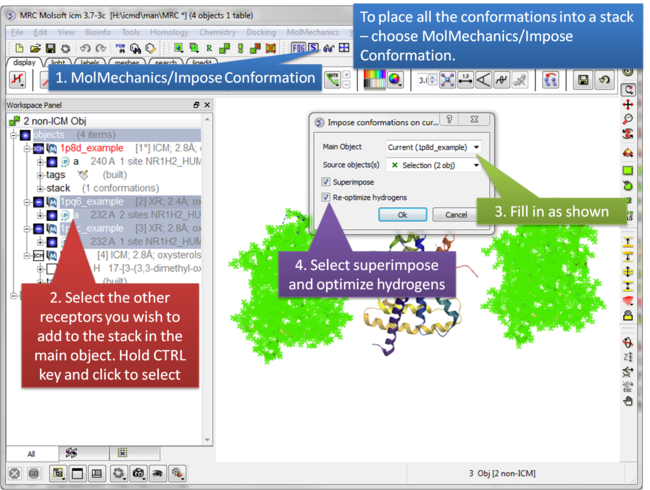 |
| Step 2: Select MolMechanics/Impose Conformation. |
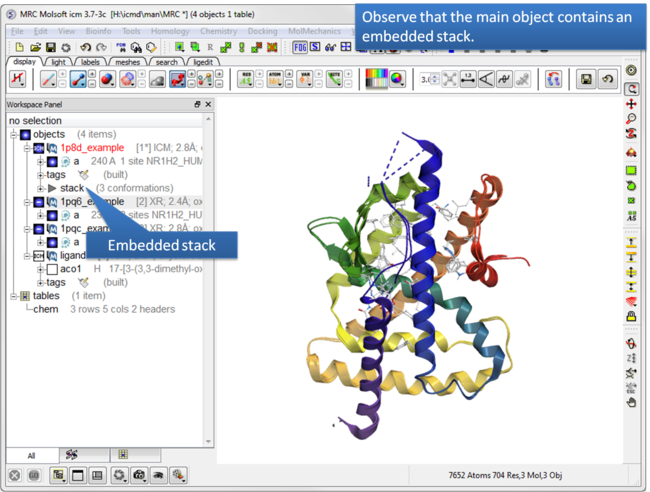 |
| Step 3: Observe the stack is embedded in the main object. |
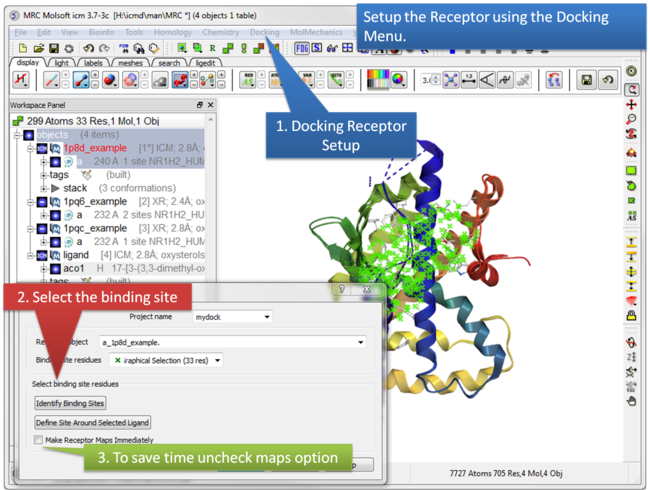 |
| Step 4:Docking/Receptor Setup . |
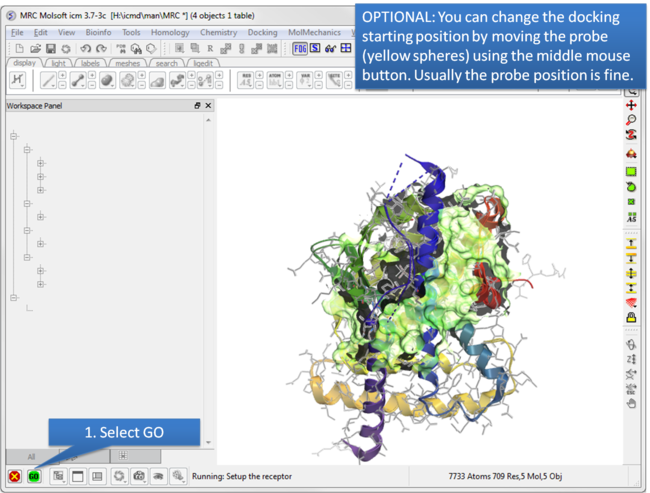 |
| Step 5: Optional: Change simulation starting point. |
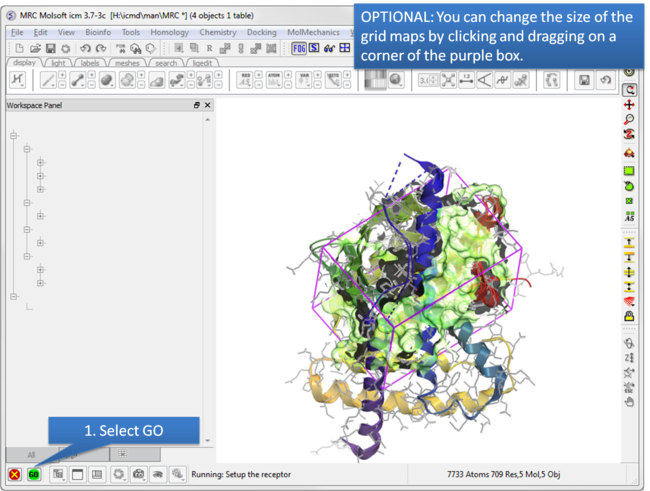 |
| Step 6: Optional: Change map size. |
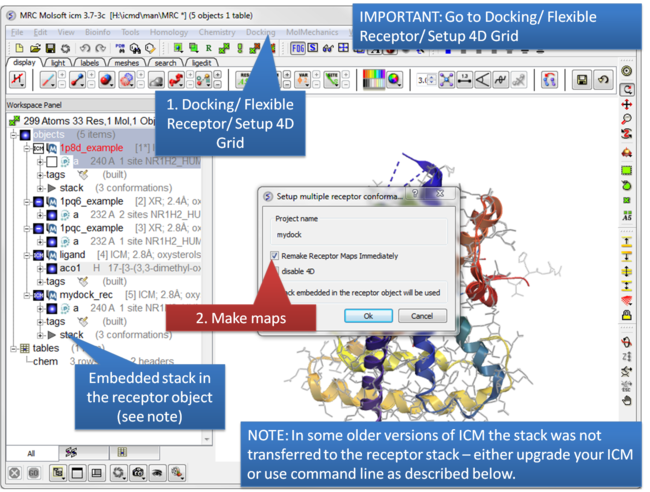 |
| Step 7: IMPORTANT: Setup the 4D maps Docking/ Flexible Receptor/ Setup 4D Grid. |
NOTE: In ICM version 3.7-3b and below it was necessary to move the stack from the main object to the receptor object. To do this:
|
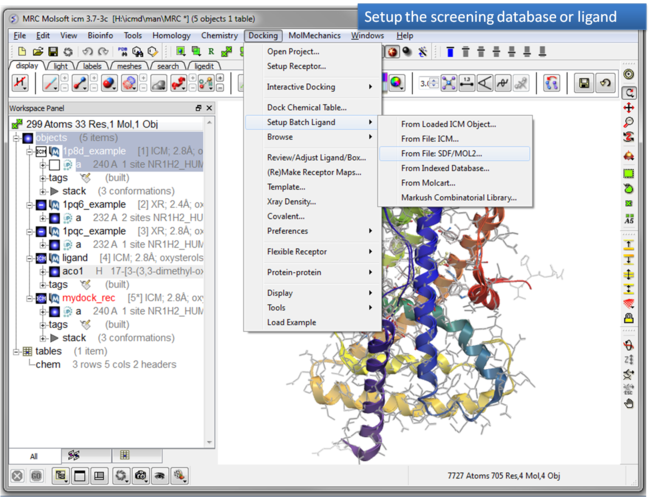 |
| Step 8: Setup the screening database. |
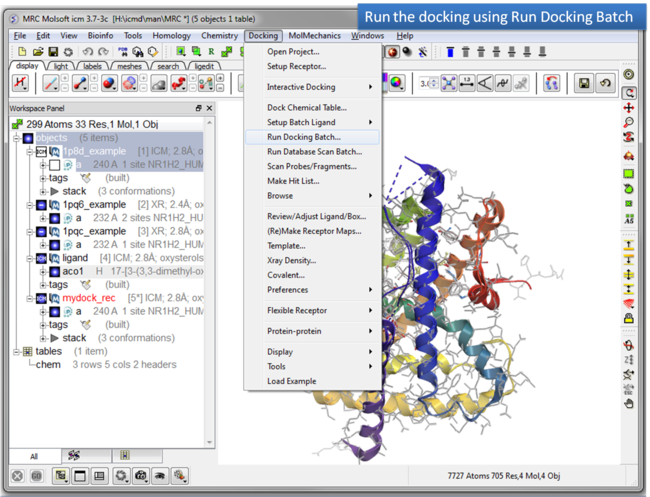 |
| Step 9: Run the docking. |
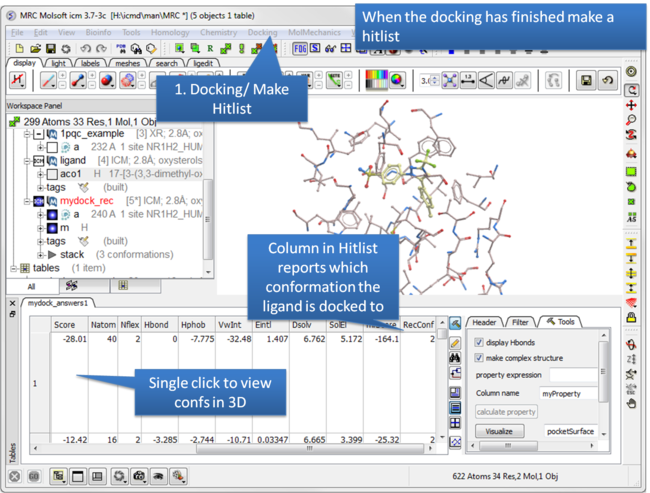 |
| Step 10: Make a hitlist. |
| Additional Resources: Tutorial |
This option allows you to keep certain residues as explicit during grid docking. For example Hydroxyls of Ser, Thr, and Tyr can be treated explicitly during docking.
- Setup the docking as described in the Small Molecule Docking section.
- Before making maps choose Docking/Flexible Receptor/Explicit Groups
The SCARE method allows you to take into account induced fit of the receptor upon ligand docking. The method systematically scans pairs of neighboring side-chains and replaces them with Alanine and docks a ligand to each of the "gapped" models. A full description of this method and validation is published here. To run the method you will need a single ligand-receptor complex. In the example below we have two JNK3 ligands, one ligand a984 cannot bind at the ATP binding pocket hinge region due to the orientation of M146. The example shows that the pocket can be opened up using SCARE to allow a984 to dock correctly.
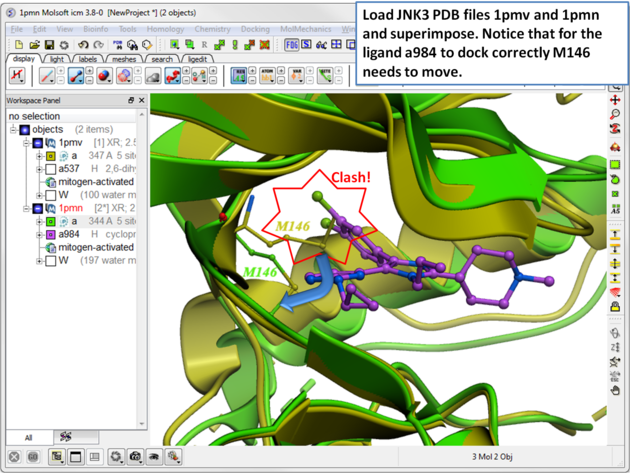 |
| Step 1 In this example we have two JNK3 receptor PDB files. To load them go to the Search tab and enter 1pmv and 1pmn. Select each receptor and then superimpose. Notice that for the ligand a984 in PDB 1pwn to dock correctly M146 needs to move. The SCARE method should be able to open up the pocket so that a984 ligand can dock correctly. |
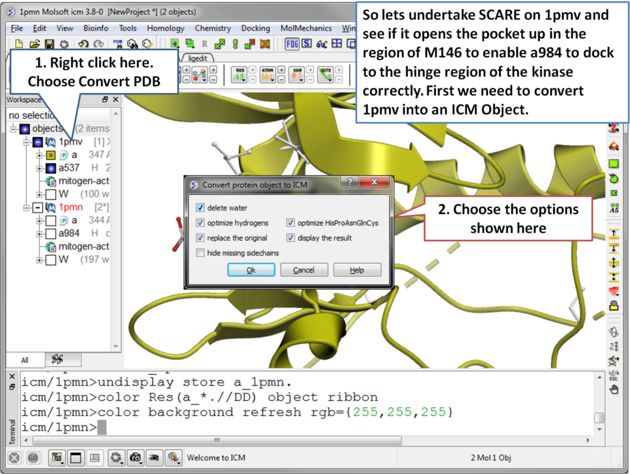 |
| Step 2 Convert 1pmv to an ICM object. |
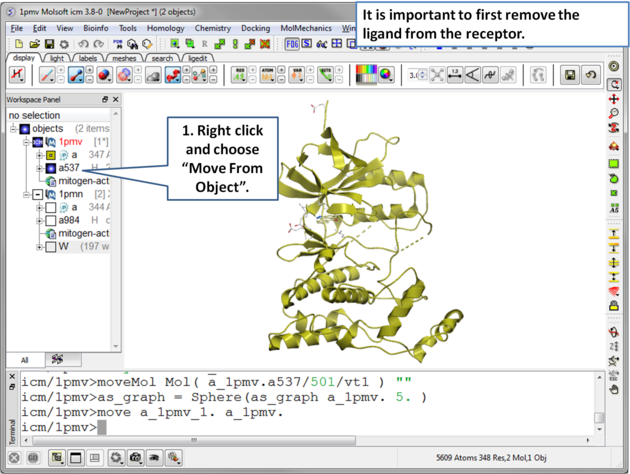 |
| Step 3 Move the ligand out of the 1pmv structure. This prevents the ligand being included in the maps. |
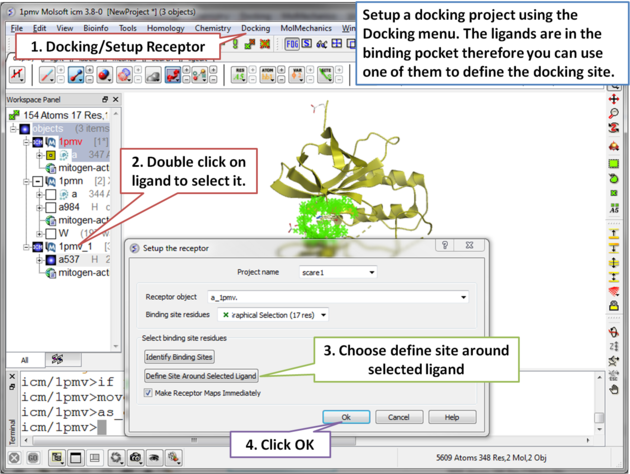 |
| Step 4 Setup the docking project by choosing the menu option Docking/Setup Receptor. |
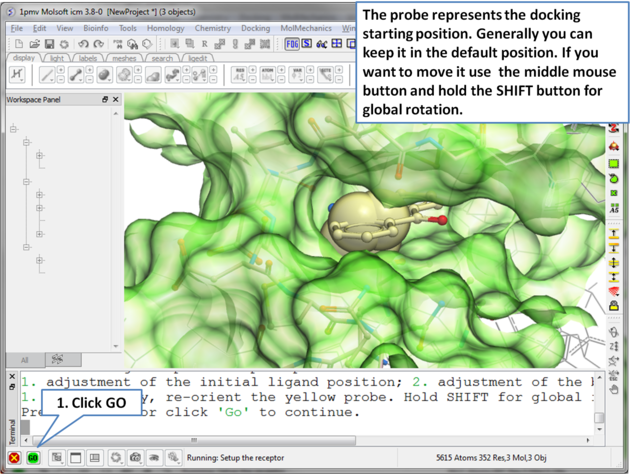 |
| Step 5 The probe represents the starting point for the docking simulation. By default the probe is in the center of the pocket. |
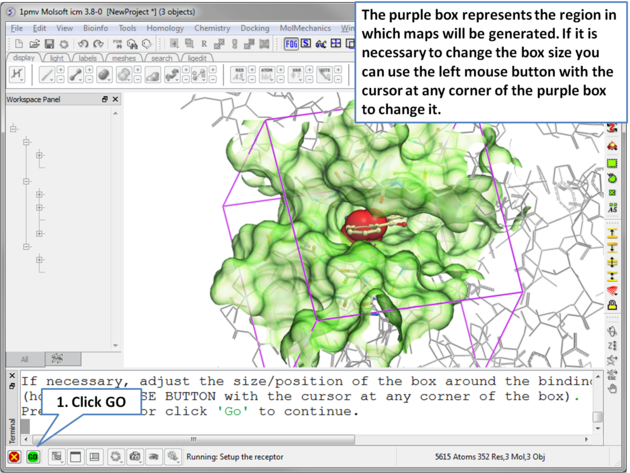 |
| Step 6 The dimensions of the box can be changed. . |
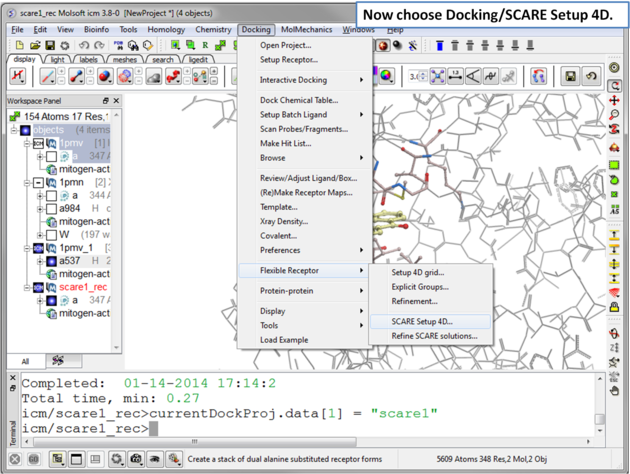 |
| Step 7 Choose the menu option Docking/Flexible Receptor/Scare Setup 4D. |
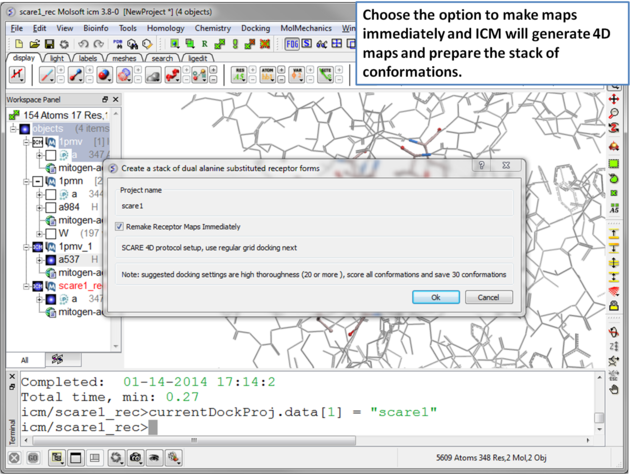 |
| Step 8 A dialog window will be displayed - click OK. |
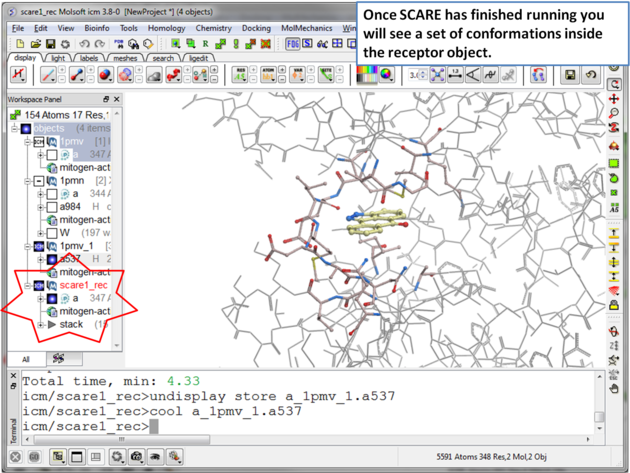 |
| Step 9 Once the SCARE simulation has finished running you will see a stack of receptor conformations in the ICM Workspace. |
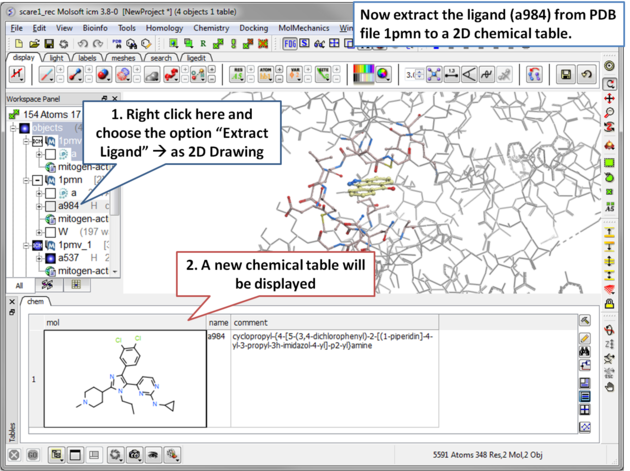 |
| Step 10 Now in preparation for ligand docking we will extract the ligand a984 from the PDB file 1pmv in the ICM Workspace. |
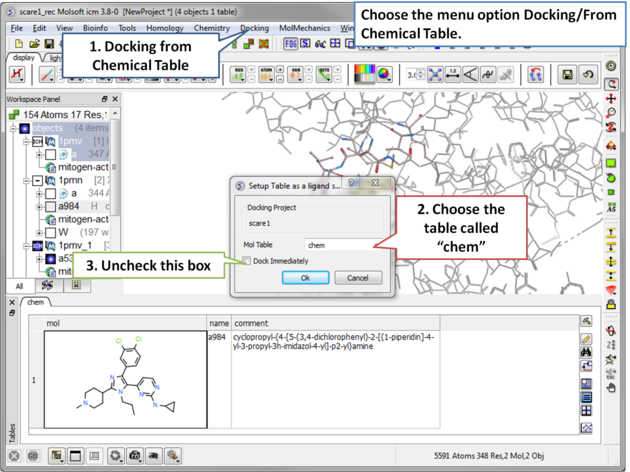 |
| Step 11 The ligand will be docked directly from the table. |
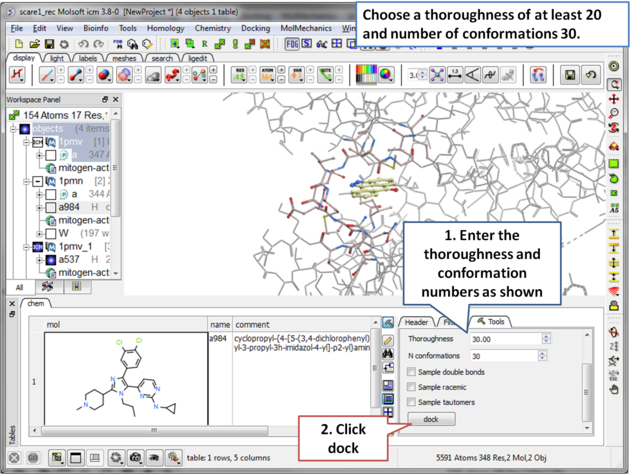 |
| Step 12 Choose a thoroughness of at least 20 and number of conformations 30. . |
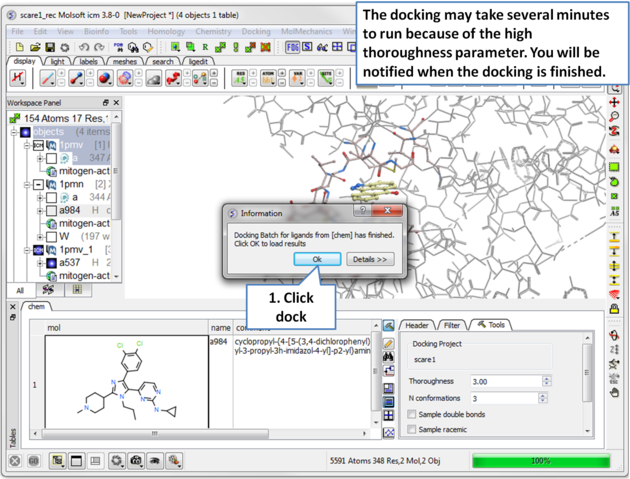 |
| Step 13 Run the docking, it may take a few minutes because the thoroughness value is high. |
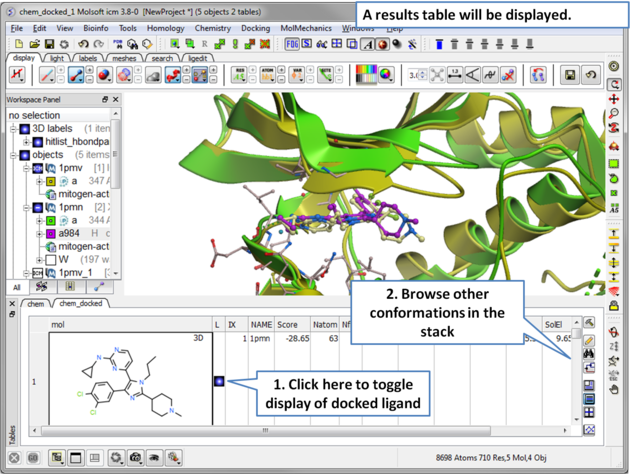 |
| Step 14 A results table will be displayed at the bottom of the gui when the docking has finished. |
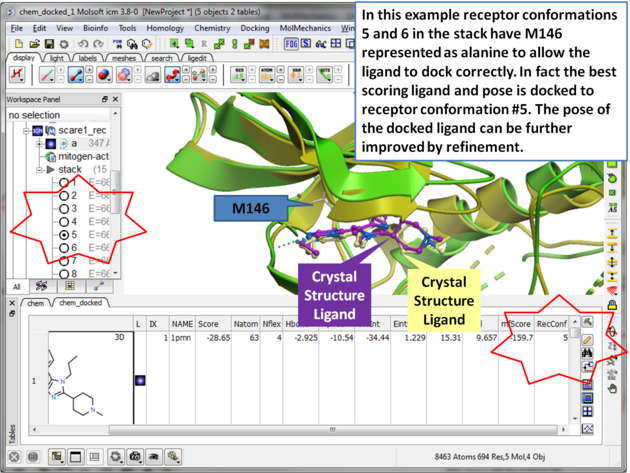 |
| Step 15 Toggle through the different conformations and observe the best pose and receptor conformation. |
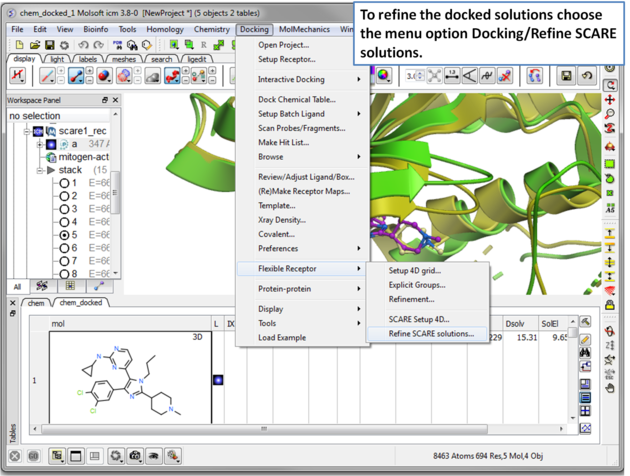 |
| Step 16 The docked ligand usually requires refinement. |
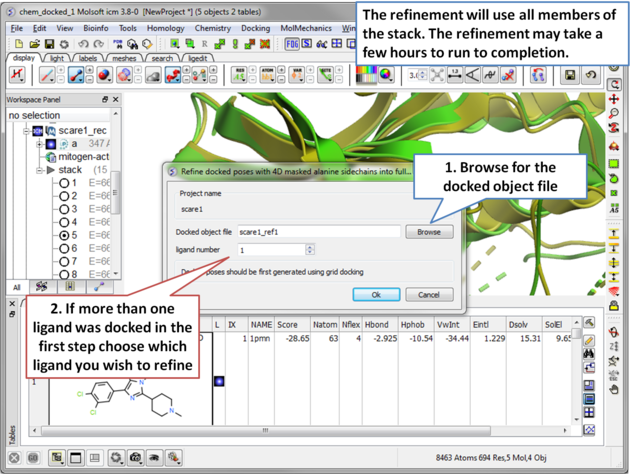 |
| Step 17 The refinement will use all members of the stack. The refinement may take a few hours to run to completion. . |
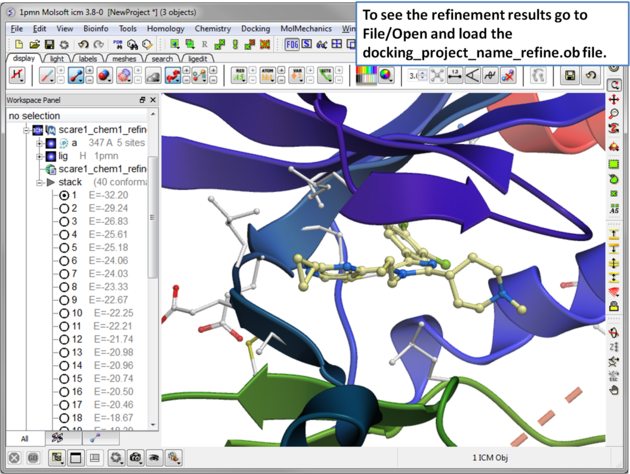 |
| Step 18 To see the refinement results go to File/Open and load the docking_project_name_refine.ob file. |