| Prev | ICM User's Guide 10.23 Generate 3D Conformers | Next |
| Available in the following product(s): ICM-Chemist-Pro | ICM-VLS |
To generate a series of conformers for a ligand(s):
- Select the compounds (row(s)) you wish to generate conformers for in an ICM Molecular Table . Or to convert a whole table of compounds select Chemistry/Generate 3D Conformers menu.
- Right click on the selected row(s) and Chemistry/Conformation Generator (selected rows) and a data entry window as shown below will be displayed.
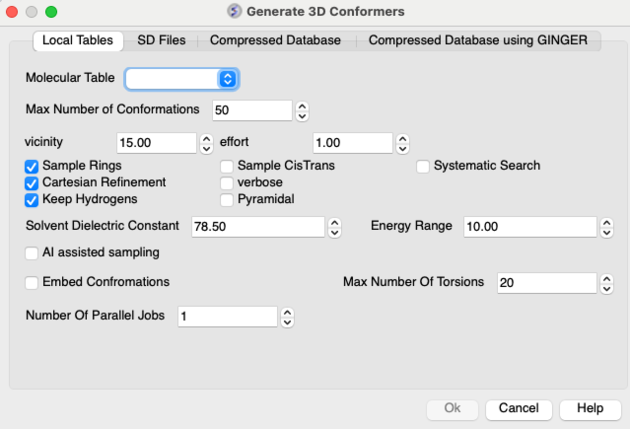
- Enter the maximum number of conformations you wish to generate.
- Enter a vicinity value. For more information on vicinity please see the command language manual http://www.molsoft.com/man/reals.html#vicinity. Density of conformers is controlled by the vicinity parameter. Currently it always operates in terms of torsion RMSD, default is 15 degrees angular RMSD. Note that we only generate local minima, therefore reducing vicinity below some level will typically not produce any new conformers.
- Enter a effort value ( thoroughness in releases before 3.8-2) . This relates to the length of sampling time. Effort multiplies the sampling length determined by default heuristic based on size and number of torsions.
- Check boxes are available for Sampling Rings, Systematic Search, Cartesian Refinement,Sample Cis and Trans, sample Pyramidal and Verbose (Display Warnings). The 'Cartesian' flag enables cartesian minimization/refinement of conformations (produced by torsion-only sampling). This should be checked whenever energetics is of importance. Torsion-only sampling is fast but only gives qualitative sample of accessible conformational space, relative energies of different conformers may not be accurate. The 'systematic' option enables systematic search through torsion space combined with local minimization. This option should be used for molecules with small number (1-3) of rotatable bonds. The 'pyrimidal' option will sample SP3 pyrimidal nitrogen.
- Click OK and the sampling will be undertaken in the background - see Windows/Background Jobs
- The 'AI Assisted' method uses AI predicted torsion profiles to guide the search. The approach is described in this MolSoft publication.
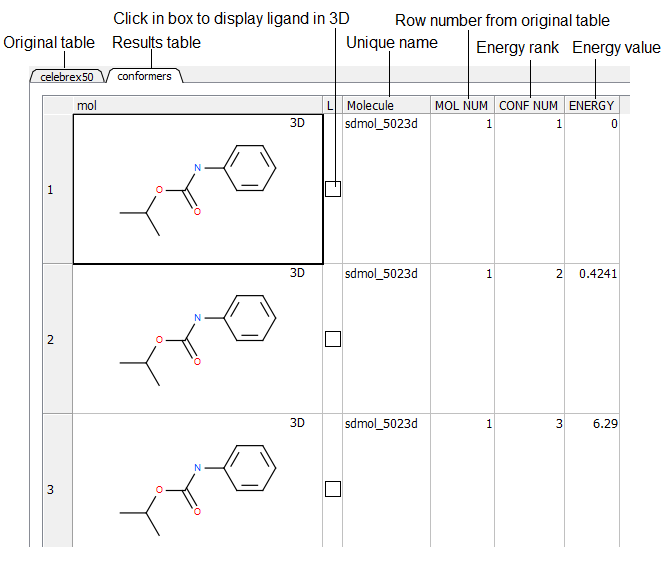
- Once the sampling has finished a table as shown below will be displayed. Click in the toggle boxes in the column labeled "L". To display more than one 3D structure at a time use the row selection options.
To generate conformers in compressed .molt format for RIDE and RIDGE.
- Go to Chemistry/Generate 3D Conformers menu.
- Select the Compressed Database or if you have a GINGER license you can use the Compressed Database using GINGER tab.
- Enter the path to your input sdf file.
- Enter the path to where you want to save the .molt file and provide a name.
- Enter maximum number of conformations you want to generate and the maximum number of torsions.
- Enter the number of parallel jobs you wish to run.
| Prev Predict | Home Up | Next Generate Tautomers |