[ Scan Hits | View Stack Conformations | Make a HitList | Hitlist Export | Reload | Displaying Docking Results ]
Docking results can be visualized and browsed in one of the following ways.
- Docking/Browse/Scan Hits - If you have docked a multi-ligand file (eg SDF) or if you want to go back and see a single docked structure you can scan the structures using this option.
- Docking/Browse/Stack Conformation - View other possible conformations for the docked ligand ranked by energy.
- Docking/Make Hit List - Rank docked compounds by ICM Docking Score Only available with ICM-VLS
The results of the docking are saved in the following files
PROJECTNAME_LIGANDNAME.ob #icm-object file with best solutions for each ligand
PROJECTNAME_*.cnf #icm conformational stack files with multiple docked conf.
The results of the docking job using ICM-VLS (separate license required) are saved in the following files:
PROJECTNAME_answers*a.ob #icm-object file with best solutions for each ligand
PROJECTNAME_*.cnf # icm conformational stack files with multiple docked conf.
PROJECTNAME_*.ou # output file where various messages are stored eg.SCORE
- Select Docking/Browse/Scan Hits and a data entry box as shown below will be displayed.
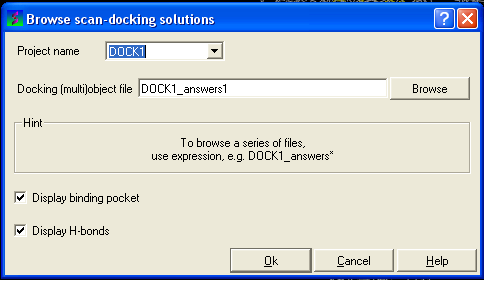
- Select the correct project name for the docking simulation results you wish to browse.
- Enter the name of the icm object file in the Docking (multi)object data entry field. This file will be called PROJECTNAME_answers*.ob or LIGAND_NAME.ob The browse button can be used to search for the correct file.
- You can display the binding pocket or the H-bonds by selecting the appropriate boxes in the Browse scan-solutions data entry window (shown above).
- Use the buttons at the bottom of the graphical user interface to browse the docked conformations. NEXT(or type "n"), BACK (or type "b"), JUMP (or type "j"), RETAIN (or type "r"), STOP (or type "s"), KEEP_STOP (or type "k").
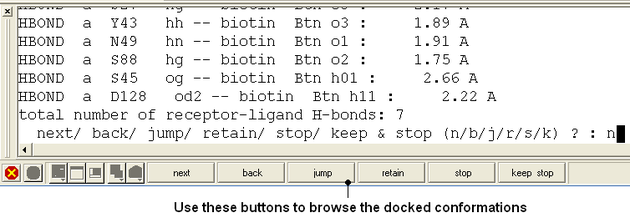
- The options keep and stop and retain will retain the displayed ligand in the graphical user interface. If you want to export the docked complex as a PDB file you will need to move the ligand and receptor into one ICM object. Moving objects is described in the FAQ section entitled How can I merge two objects into one?
To view the multiple positions of a single ligand in the docking simulation ranked by energy.
- Select menu Docking/Browse/Stack Conformations
The Browse Stack Conformation data entry window will be displayed.
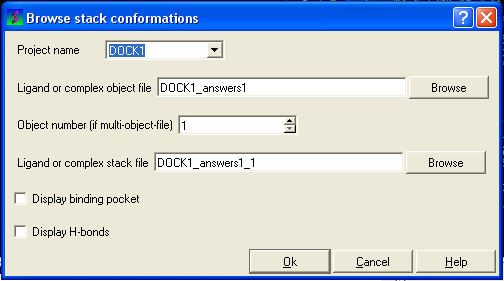
- Select the correct project name for the docking simulation results you wish to browse.
- Enter the name of the icm object file in the Docking (multi)object data entry field. This file will be called PROJECTNAME_answers*.ob .The browse button can be used to search for the correct file.
- Enter the name of the icm conformational stack files with multiple docked conformations into the Ligand or complex stack file data entry box. This file will be called PROJECTNAME1_1.cnf . The browse button can be used to search for the correct file. The second solution in the stack can be viewed by changing the number 1 at the end of the file name to 2 (PROJECTNAME1_2.cnf) and so on for each solution in the stack.
- You can display the binding pocket or the H-bonds by selecting the appropriate boxes in the Browse scan-solutions data entry window (shown above).
- Use the buttons at the bottom of the graphical user interface to browse the docked conformations. NEXT(or type "n"), BACK (or type "b"), JUMP (or type "j"), RETAIN (or type "r"), STOP (or type "s"), KEEP_STOP (or type "k").
- The options keep and stop and retain will retain the displayed ligand in the graphical user interface. If you want to export the docked complex as a PDB file you will need to move the ligand and receptor into one ICM object. Moving objects is described in the FAQ section entitled How can I merge two objects into one?
Columns in the Stack Table
i rank in stack
ener Energy kcal/mol
gvw van der Waals grid potential
gb hydrogen bonding grid potential
ge electrostatic grid potential
gs hydrophobic grid potential
Einternal is internal conformation energy of the ligand. This value represents the energy difference between the relaxed (unbound) ligand and its conformation in the docked (bound) state - so lower values are better. It also contributes to the physics-based docking score (the ‘Score’ value).
To calculate this, an ensemble of low-energy ligand conformers is generated in solution using the MMFF94s force field. The lowest-energy conformer is used as the unstrained reference. Strain is then defined as the energy difference between this reference conformation and the docked pose, with both energies computed using the same force field.
[ Hitlist Columns ]
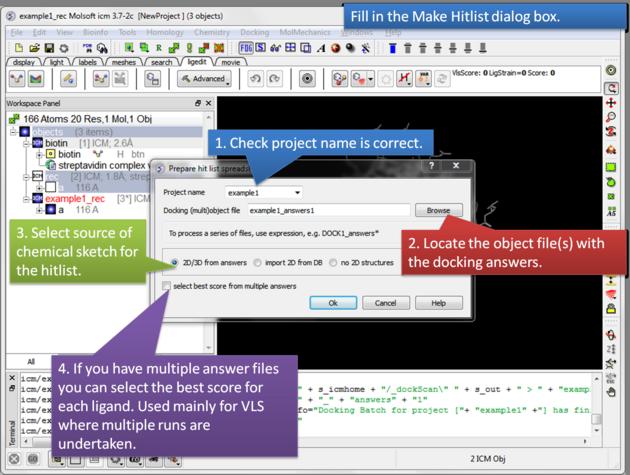
A hilist is a convenient way to view your docking results in a chemical spreadsheet. To make a hitlist
- Docking/Make Hit List
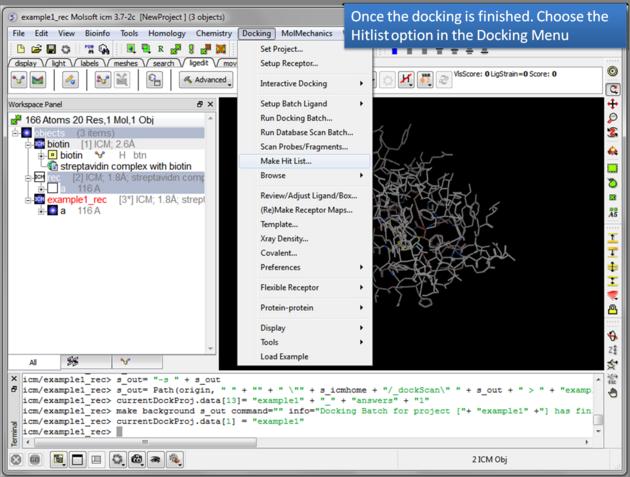
- Enter project name
- Use the browse button to locate DockingProjectName_answers.ob file
- You can include a 2D image into the HITLIST
- Select Unique if you have made multiple docking runs the best docking score will be taken to make the hitlist unique.
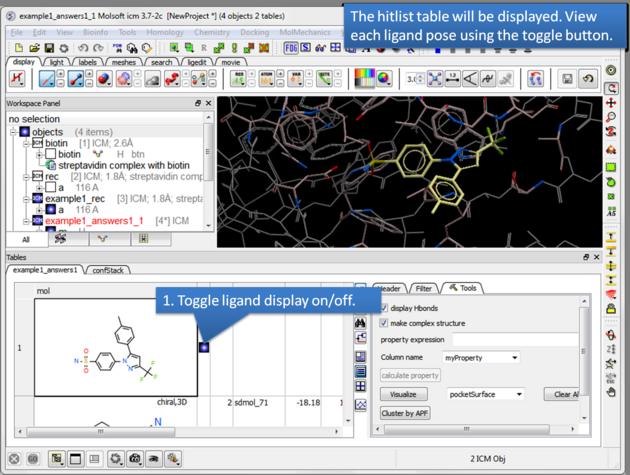
- A HITLIST table will be displayed. Each docked ligand can be viewed by double clicking in the HITLIST table. A stack of conformations for each ligand will also be displayed in a table.
There is a toggle button to easily undisplay or display each ligand pose.
Columns in the HitList Table with ICM-VLS LICENSE
- IX is the index number from the docked database
- Score is the ICM score -32 and lower are generally considered good scores - but depends on the receptor (e.g. exposed pockets or pockets with metal ions mayhave higher scores than -32) ( More... ).
- Natom is the number of atoms in docked ligand
- Nflex is the number of rotatable torsions
- Hbond is Hydrogen Bond energy - (lower the better)
- Hphob is the hydrophobic energy in exposing a surface to water (lower the better)
- VwInt is the van der Waals interaction energy (sum of gc and gh van der waals). Current version of the score uses explicit van der Waals interaction energy calculation (no grids). (lower the better)
- Eintl is internal conformation energy of the ligand. (lower the better). We suggest cutoff between 5 and 10, depending on how strict or permissive you want to be. More permissive settings may be justified by the noise in strain evaluation and lack of full receptor relaxation (induced fit would potentially let the ligand relax further). Also larger ligands may generally give larger strains. Eint is a component of the Score - default weight is 0.6. This value represents the energy difference between the relaxed (unbound) ligand and its conformation in the docked (bound) state - so lower values are better. It also contributes to the physics-based docking score (the Score value). To calculate this, an ensemble of low-energy ligand conformers is generated in solution using the MMFF94s force field. The lowest-energy conformer is used as the unstrained reference. Strain is then defined as the energy difference between this reference conformation and the docked pose, with both energies computed using the same force field.
- Dsolv is the desolvation of exposed h-bond donors and acceptors (lower the better)
- SolEl is the solvation electrostatics energy change upon binding (lower the better)
- dTSc Loss of entropy by the rotatable protein side-chains.
- mfScore is the potential of mean force score.
- RTCNN Score is a Neural Network Score - the lower the score the better the predicted interaction. ( More... )
- RecConf - if multiple receptor conformations was used Docking/Flexible Receptor/Setup 4D grid and represents the receptor conformation number
Columns in the HitList Table with ICM-PRO ONLY - NO ICM-VLS LICENSE
- Edoc = grid docking energy - the lower the better. It is the sum of the interaction terms reported below, plus internal energy of the ligand (~strain). Units are kcal/mol, this is am energy model intended primarily to rank well the near-native poses.
- Egb = hydrogen bond grid energy http://www.molsoft.com/man/terms.html#term-gb
- Ege = electrostatic grid potential http://www.molsoft.com/man/terms.html#term-ge
- Egs = hydrophobic potential http://www.molsoft.com/man/terms.html#term-gs
- Egv = is grid-based van der Waals (ia sum of .gh' http://www.molsoft.com/man/terms.html#term-gh and .gc' http://www.molsoft.com/man/terms.html#term-gc )
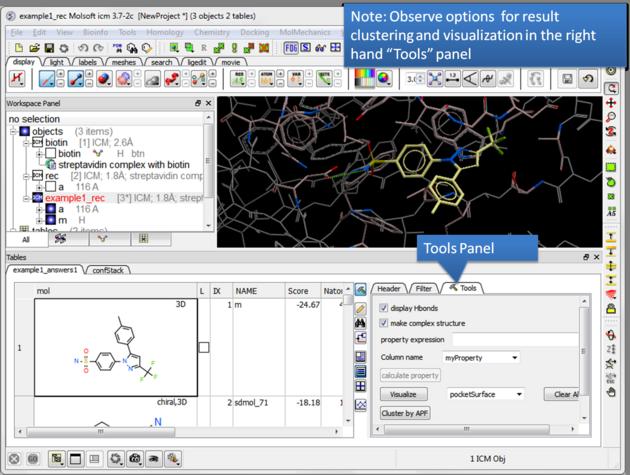
| NOTE There are a number of post-screening analysis built into the Tools panel on the right hand side of the hitlist. These are described here. |
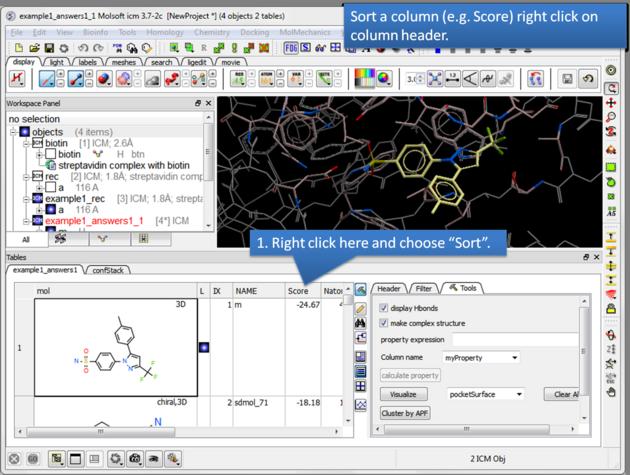 |
| How to sort the hitlist. Right click on a column header and select "Sort". For example you may want to sort by docking score. |
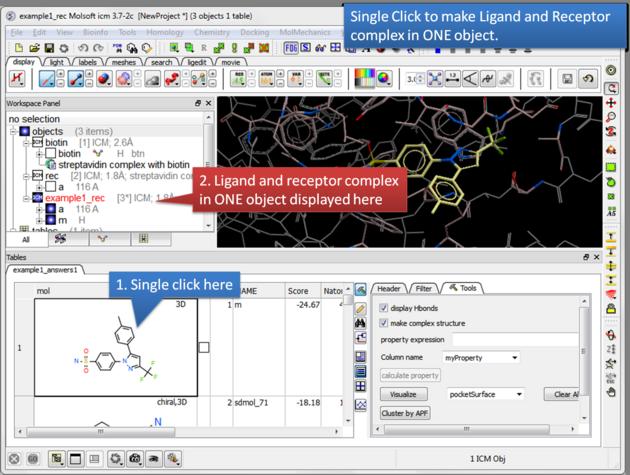 |
| Single click. A single click on a row in the hitlist will load the ligand and receptor complex in one object. |
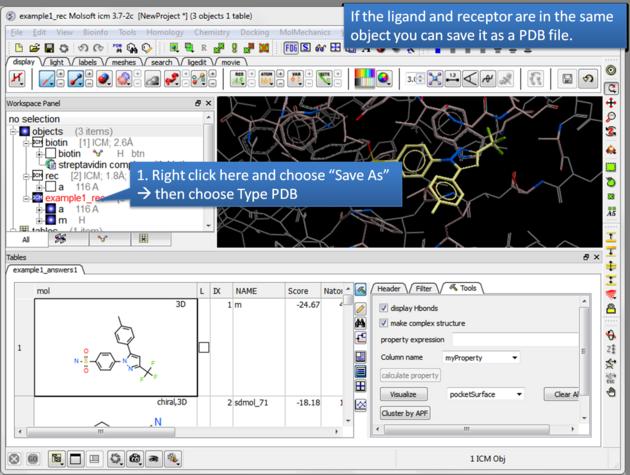 |
| Save a complex as a PDB file. If the ligand and receptor are in the same object you can save them both as a PDB file. Right click on the name of the complex in the ICM Workspace. Select "Save as" and then a Windows dialog window will be displayed. Use the drop down button to select Type PDB (.pdb .ent). |
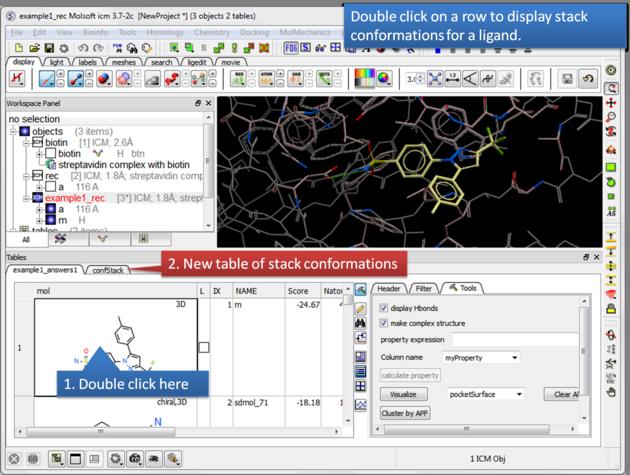 |
| Display stack of alternative ligand conformations. Double click on a row generates a new stack conformation table. |
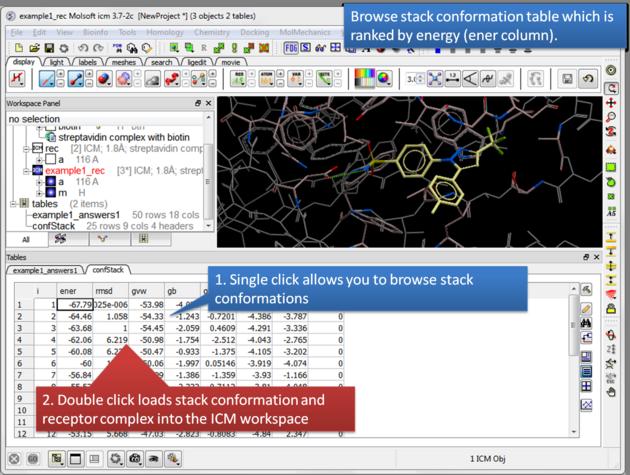 |
| Browse stack conformations. Single click allows you to browse (or use up/down arrow keys) and double click generates complex in the ICM workspace. |
 |
| Display stack of alternative ligand conformations. Double click on a row generates a new stack conformation table. |
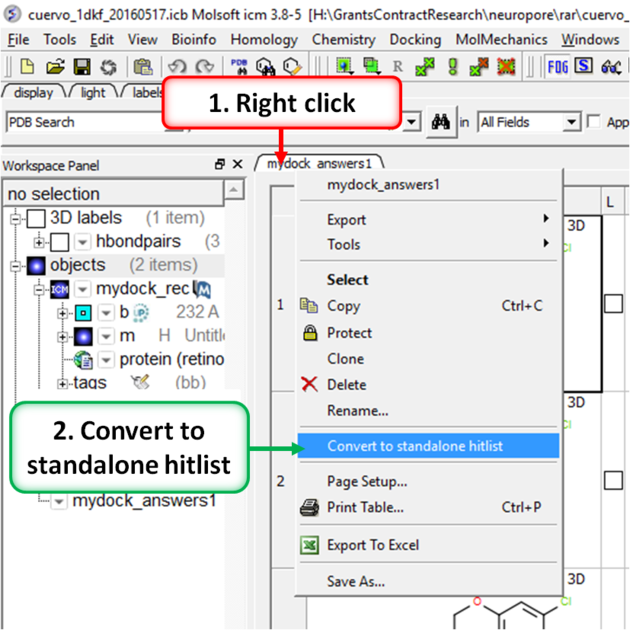
A docking hitlist can be made into a standalone docking project file to share with others and open in locations away from the docking directory. This can be done by:
- Make the docking hitlist Docking/Make Hitlist.
- Right click on the hitlist header tab (see image below).
- Select Convert To Standalone Hitlist
- File/ Save Project and save the project as an .icb file that others can view using ICM or the free ICM-Browser.
To reload a docking project in version 3.7-2 and earlier.
/Docking/Set Project - Type in the Docking Project Name (Case Sensitive)
or in versions above 3.7-2
/Docking/Open Project
Now you can browse scan solutions etc.... and use the maps to dock another ligand.
After docking you will make a hitlist and in the hitlist table there some options to help to display the ligand-receptor complex. These options are in the extra panel section of the hitlist table. Here is how to access them:
- Single click on one row of your hitlist to load the ligand (usually labeled m) into the receptor object (_rec object).
- Click on the Extra panel button on the hitlist.
- Select the Tools tab.
- Go to the Visualizations section and use the drop down button to choose "Pretty View".
- Click on the Visualizations option and the complex will be displayed in "Pretty View".
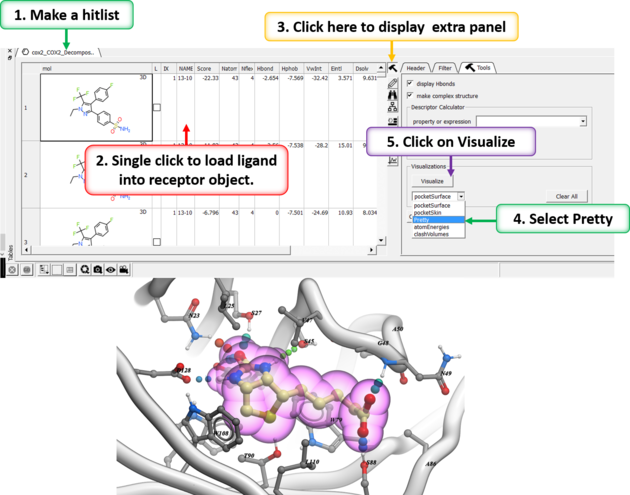
The same panel can be used to display:
- Pocket Surface a mesh representation of the ligand binding pocket colored by properties of the ligand (opposite of pocket skin below). White=aromatic lipophilic, Green=non-aromatic other (mostly aliphatic) lipophilic surface, Red=hydrogen bonding acceptor potential, Blue=hydrogen bond donor potential.
- Pocket Skin - a surface colored by the binding properties of the pocket. White=aromatic lipophilic, Green=non-aromatic other (mostly aliphatic) lipophilic surface, Red=hydrogen bonding acceptor potential, Blue=hydrogen bond donor potential.
- Atom Energies - contribution of each atom to the Docking Score.
- Clash Volumes - highlights any clashes between ligand and receptor.
| Note There are additional display preferences you can set using Docking/Preferences/Display. |