Google Search: Keyword Search:
| Prev | ICM User's Guide 4.1 Convert to ICM Object | Next |
[ Load a Protein Structure | Convert ]
| Available in the following product(s): ICM-Browser | ICM-Browser-Pro | ICM-Pro | ICM-Chemist |
4.1.1 Load a Protein Structure |
There are a couple of ways to read into ICM a protein structure.
- Search the PDB using the Search Tab.
- Load in a PDB file that you have saved on your computer using File/Open.
4.1.2 Converting PDB Files Into ICM Objects |
If you are going to make any type of energy calculation in ICM (eg docking, display H-bonds, display electrostatic and binding property surfaces etc..) it is necessary to convert a protein or chemical into an ICM object. Be aware that upon conversion ICM adds missing side-chain atoms (but wont try to build missing loops) due to the nature of the internal-coordinate system. The list of residues/ atoms added is presented in the command line shell and can be reviewed. For reference, the original PDB entry is kept in the system. See the command line manual for a more complete description of what the conversion process does.
| NOTE: Before converting a protein structure to an ICM object make sure that the chemicals contained within the structure (e.g. ligands) are correct. If an error is found you can edit the ligand as described here. |
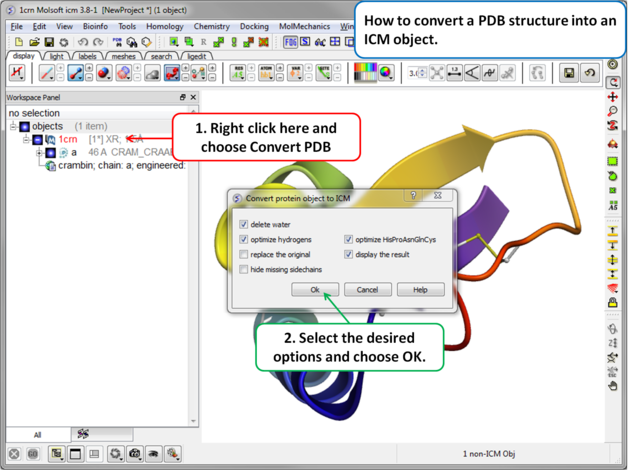
To convert a PDB structure into an ICM object follow the steps shown below:
- Right click on the name of the protein you wish to convert in the ICM Workspace.
- A dialog box will be displayed as shown below.
- To delete the water molecules select Delete Waters, loose waters are defined as those not making any h-bonds with the ligand or protein, tight waters are those making 3 or more h-bonds.
- To optimize hydrogen atoms (recommended) select Optimize Hydrogens. This option performs global optimization of hydrogens to find the best hydrogen bonding network.
- To optimize the orientation of His, Pro, Asn, Gln, Cys residues then choose optimizeHisProAsnGlnCys. The following residues will be further optimized: His - three protonation states and two rotations will be tried and the residue will be renamed according to its subtype: hie (epsilon tautomer) or hip (+). Asn and Gln - (a 180 deg. flip will be tried). Cys - in the vicinity of Zn, Cu, Fe and Co to cym.
- To keep a copy of your PDB file uncheck the option replace original.
- The converted structure can be displayed immediately by checking display the result
- Uncheck the box hide missing side chains if you want ICM to build missing heavy atoms that are not reported in the PDB (due to the lack of density), they will be added according to the residue name and assigned zero occupancies. Check this box if you want residues missing heavy atoms to be hidden.
- Tautomer Optimize ligand tautomer. For each ligand (hetero a_H), different tautomeric forms are generated and their energies are evaluated using the “hb, vw, 14, el” terms in the context of other chains. The lowest-energy tautomer is then selected as the preferred form.
If your object is an ICM object it will display ICM next to the molecule in the ICM Workspace.

| Prev Protein Structure | Home Up | Next Pocket Display |