[ Sketch Markush Structure | R-Group | Create Markush | Enumerate by Scaffold | Enumerate by Reaction ]
| Available in the following product(s): ICM-Chemist | ICM-Chemist-Pro | ICM-VLS |
To create or modify a Markush Structure:
- Use the Molecular Editor to sketch the scaffold as shown below. Right click on an atom and choose -> Element-> R1,R2....
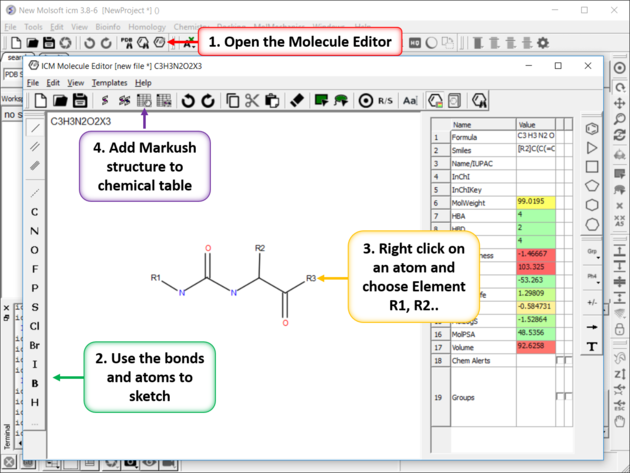
- Save the sketch to a chemical table by clicking on the "Append Molecule to Chemical Table" button in the Molecule Editor.
- Close the Molecular Editor window by clicking on the cross in the top right hand corner and the changes will be submitted to the table.
ICM has an inbuilt local database of R-groups generated from ChEMBL data. To open this file:
- Chemistry/Combinatorial Chemistry/ Load R Group Database
- A table of R-groups will be displayed - attachment points are represented by asterisk. The 'freq' column represents the number of pairs found in ChEMBL for that scaffold.
You can sketch your own substituents:
- Open the Molecular Editor.
- Sketch the substituent and use
>right click>attachment point to assign the attachment point.
You can also split a chemical table into fragments as described here.
You can read in an SDF, mol, smiles file containing fragments. If you do not want the first atom of the substituents to be the attachment point you need to define the attachment point. Attachment points are automatically assigned when you extract fragments or you can define them manually by using the molecular editor (as described above).
In many combinatorial chemistry options there is an option to use LigEdit Mofifiers. These are the substituent groups you see in the edit panel in the 3D Ligand editor.
To create a Markush structure:
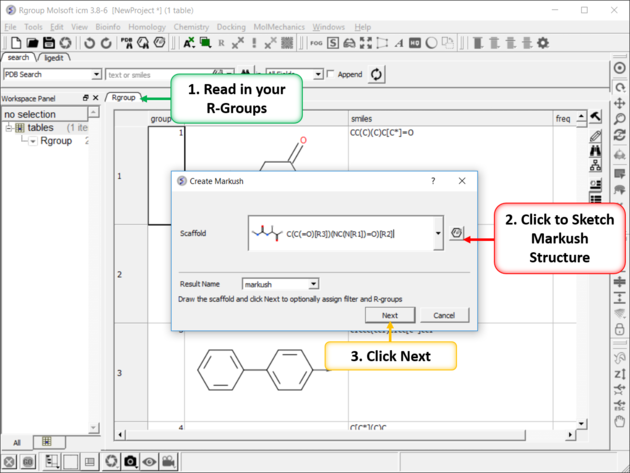 |
 |
|
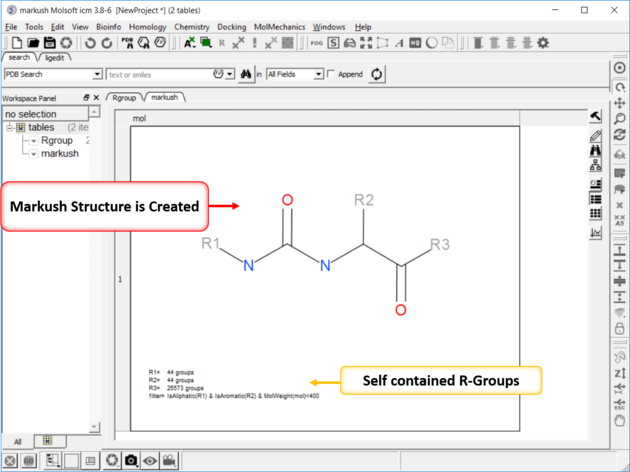 |
|
| NOTE: Once a Markush is created you can right click on it in the chemical table you will see two convenient options 1. Edit/Markush Scaffold Properties and 2. Extract Markush Scaffold R-groups. |
To enumerate a library based on R-groups:
- Sketch the Markush structure and save it in a chemical spreadsheet.
- Read in the R-Group substituents you wish to use to enumerate the library.
- Right click on the structure and select Chemistry/Enumerate R-groups or use the Chemistry/Combinatorial Chemistry/ Enumerate by Scaffold-Markush menu. If you use the menu option you will need to choose the table containing the scaffold from the drop down list of currently loaded tables. The index number refers to the row number in the scaffold table. Click OK.
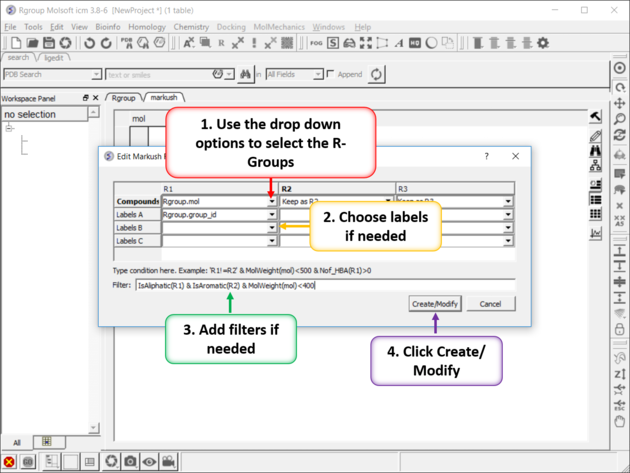
- Select the R1, R2... table , labels and filters if necessary. As an example of a filter you could use R1 !=R2, OR MolWeight<500, OR IsAliphatic(R1) & IsAromatic(R2) & MolWeight(mol)<400. Currently supported filters include: Normalize,Real,Integer,String,Min,Max,Mean,Rmsd,Sum,Ceil,Floor,Sign,Log,Sqrt, Power,Split,Nof,MolWeight,MolFormula,IupacName,Nof_Molecules,Nof_Atoms,Nof_Fragments,Nof_Chirals,Nof_RotB,Nof_HBA, Nof_HBD,Nof_Rings,Max_Ring_Size,Min_Ring_Size,Max_Fused_Rings,IsAromatic,IsAliphatic,Smiles,MolLogP, MolLogS,MolVolume,MoldHf,MolPSA,MolArea,DrugLikeness,MolhERG,MolHalfLife,MolPAINS,MolCharge,NofSites.
- A new table will be produced called T_enum with the Template structure highlighted in red.
| Additional Resources: Tutorial | Video | Command Line Docs |
Reactions can be drawn using the ICM Molecular Editor. Reactants should be drawn side-by-side (no + sign is necessary) and separated from the product using the arrow. See example shown below:
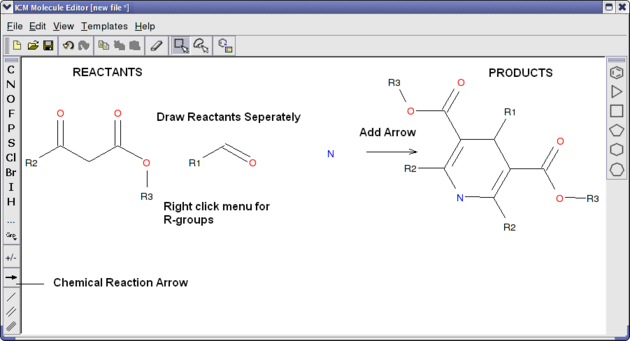
This example is available in the ICM distribution as example_reaction1.icb. The reaction is the Hantzsch Dihydropyridine (Pyridine) Synthesis. This reaction allows the preparation of dihydropyridine derivatives by condensation of an aldehyde with two equivalents of a beta-ketoester in the presence of ammonia. Subsequent oxidation (or dehydrogenation) gives pyridine-3,5-dicarboxylates, which may also be decarboxylated to yield the corresponding pyridines.
In this example we have two reactants therefore it is necessary to have two reactant substructure tables loaded into ICM. ICM will match the substructure drawn in the reaction with the chemicals in the reactant table.
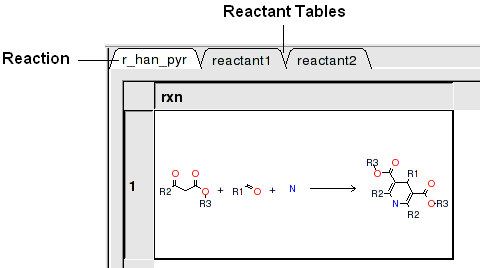
reactant 1 table:
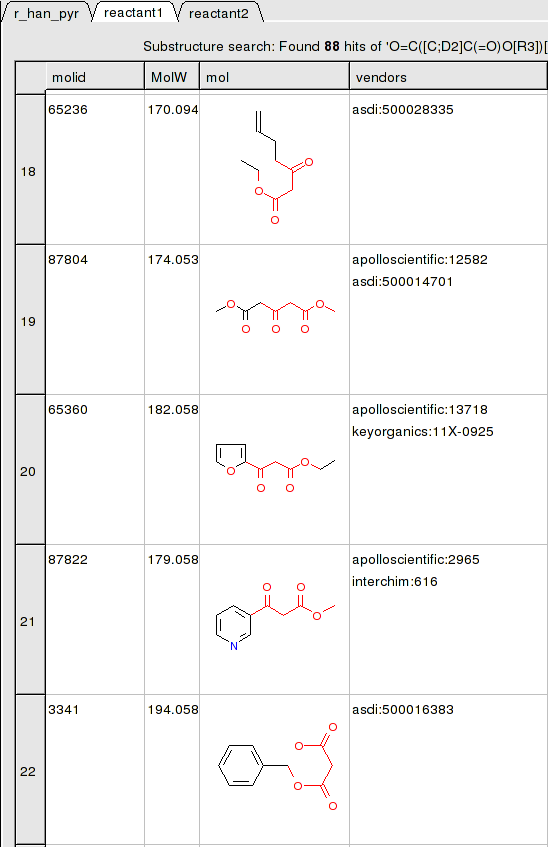
To apply a reaction:
- Chemistry/Enumerate by Reaction.
- In this example (example_reaction1.icb) we already have the reaction drawn in a chemical table. Therefore select the Choose Table With Reaction. If you would like to draw a new reaction select Draw New Reaction.
- Enter the name of the table containing the reaction. If you have more than one reaction drawn you can select the row using the index option.
- Click OK and then you will be asked to enter the Reactants. Select the reactant tables from the drop down arrow for Reactant 1 and Reactant 2.
- You can transfer information to the reactant table by selecting columns in the Labels section.
- Unused reactants can be marked.
- Select what you want to do with multiple matches. The option 'skip' will skip compounds where there are more than one way to match the fragment and 'first' will take first match and ignore others.
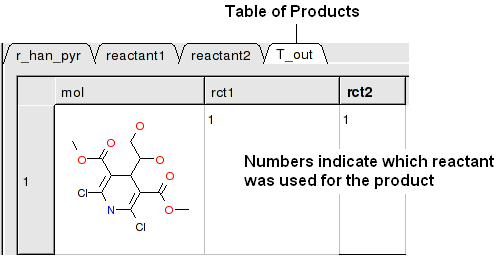
A table of Products will be then displayed in a table called T_out. Columns in T_out labeled "rct" display which reactants were used to build the product.