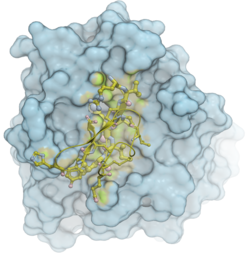
In this example we will use the Fast Fourier Transform (FFT) docking method to re-dock a trypsin inhibitor (pdb: 1lu0) to trypsin (pdb: 1btp) and compare the docked molecules with the complex (pdb: 1ppe). The docking method contains two stages:
- The first stage uses a simplified scoring function representing steric fit and hydrophobic/hydrophilic contact matching. FFT is then used for translational search using systematic search of rotations from 60x27 (coarse) to 256x125 (fine) orientations.
- The second stage rescores the top 3000-20000 solutions with a more accurate energy function including electrostatics and SAS-based solvation. The conformations are then clustered using contact fingerprints.
Selected complexes in the stack of hits can then be refined with flexible side-chains.
The steps for each stage of the docking are described here:
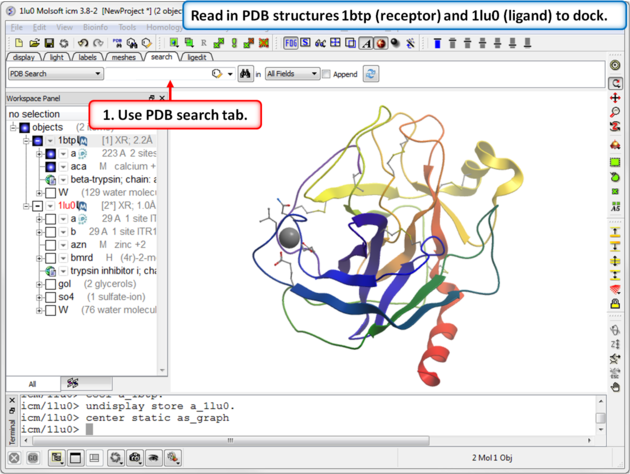 |
| Step 1: Read in PDB structures 1btp (receptor) and 1lu0 (ligand) to dock. |
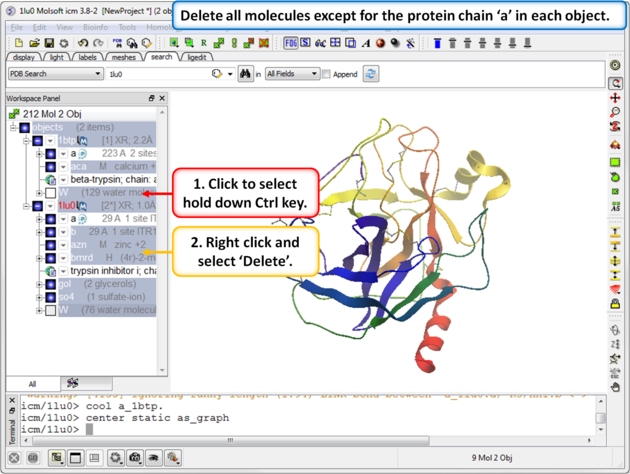 |
| Step 2: Delete all molecules except for the protein chain "a" in each object. |
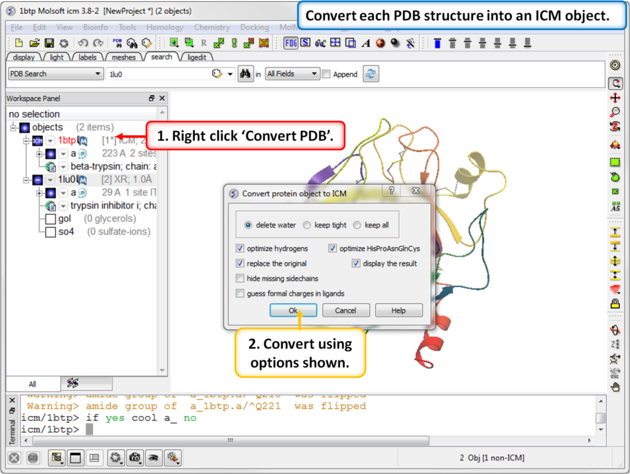 |
| Step 3: Convert each PDB structure into an ICM object. |
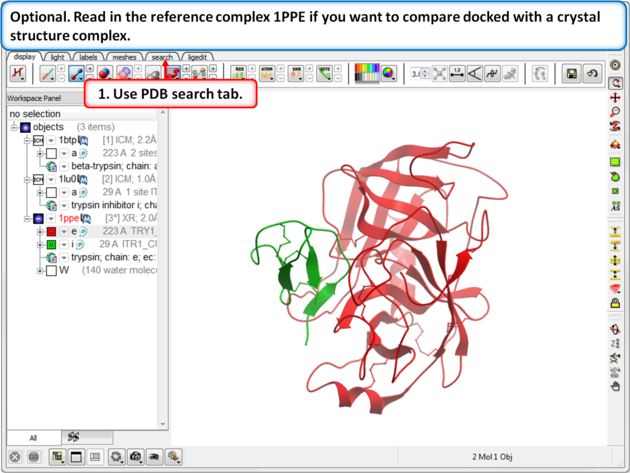 |
| Step 4: Optional. Read in the reference complex 1PPE if you want to compare docked with a crystal structure complex. |
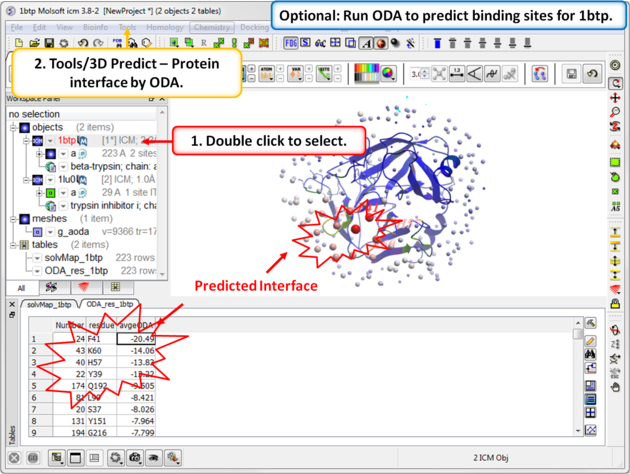 |
| Step 5: Optional. Run Optimal Docking Area (ODA) to predict binding sites for 1btp. |
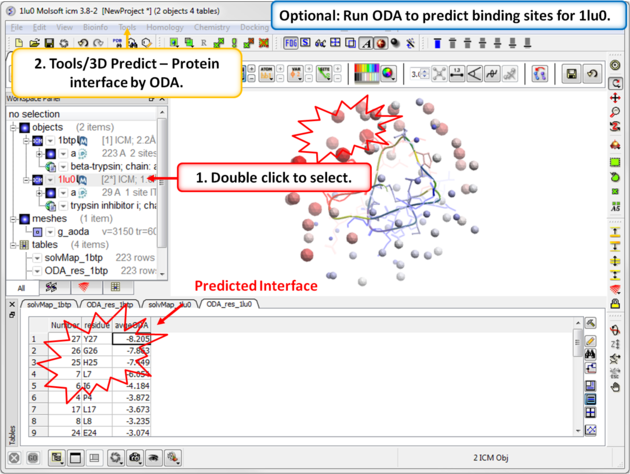 |
| Step 6: Optional. Run Optimal Docking Area (ODA) to predict binding sites for 1lu0. |
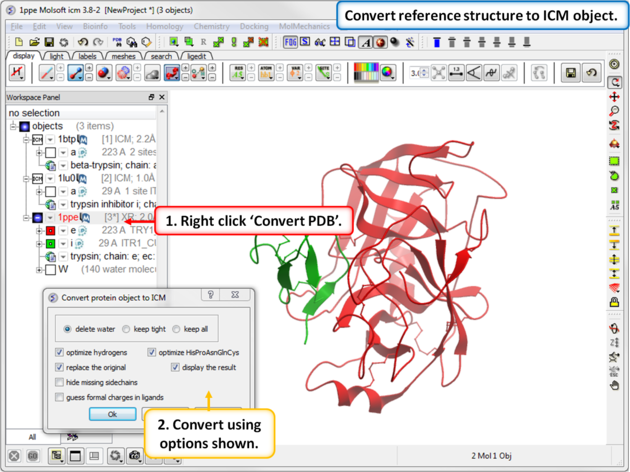 |
| Step 7: Convert reference structure to ICM object. |
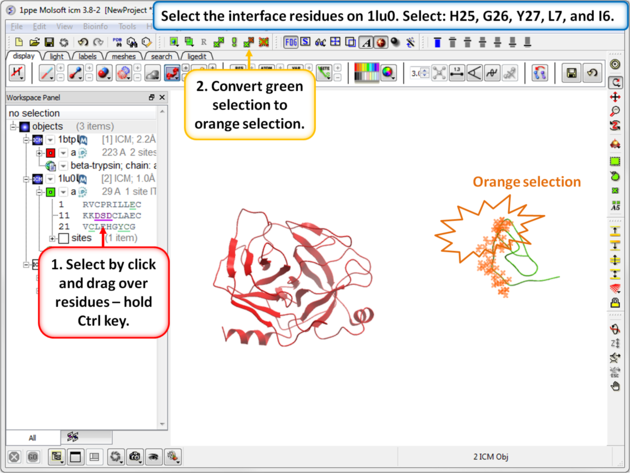 |
| Step 8: Select the interface residues on 1lu0. Select: H25, G26, Y27, L7, and I6. Convert the green selection to an orange selection. |
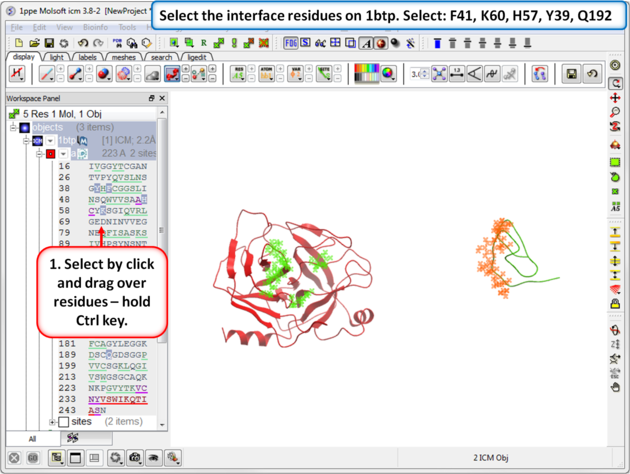 |
| Step 9: Select the interface residues on 1btp. Select: F41, K60, H57, Y39, Q192 |
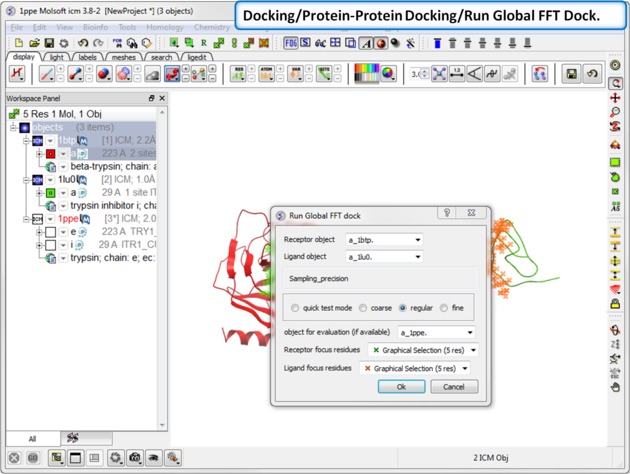 |
| Step 10: Start the docking go to Docking/Protein-Protein Docking/Run Global FFT Dock. |
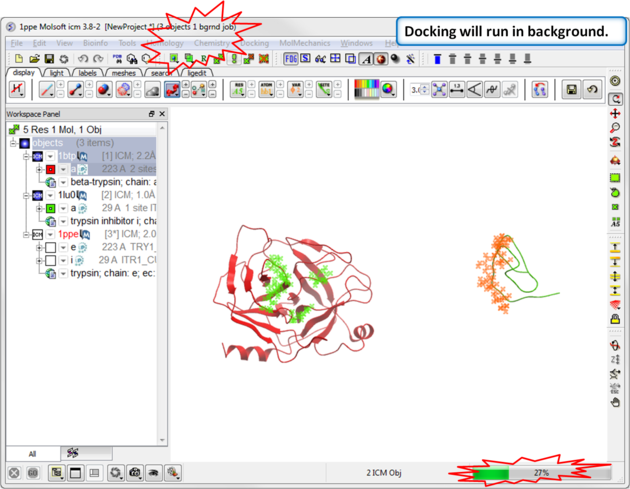 |
| Step 11: Docking will run in the background, you can monitor this by going to Windows/Background Jobs. |
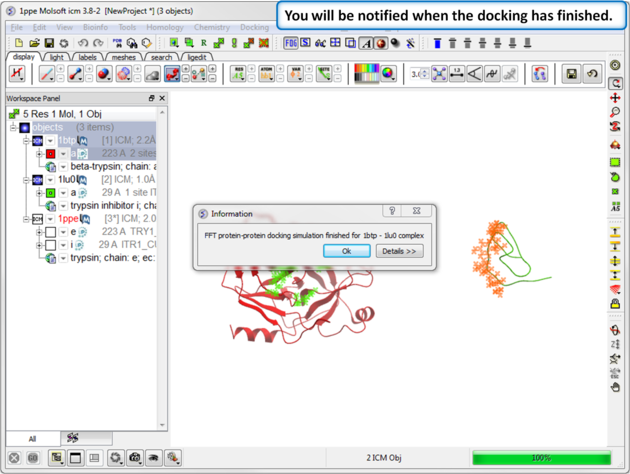 |
| Step 12: You will be notified when the docking has finished. |
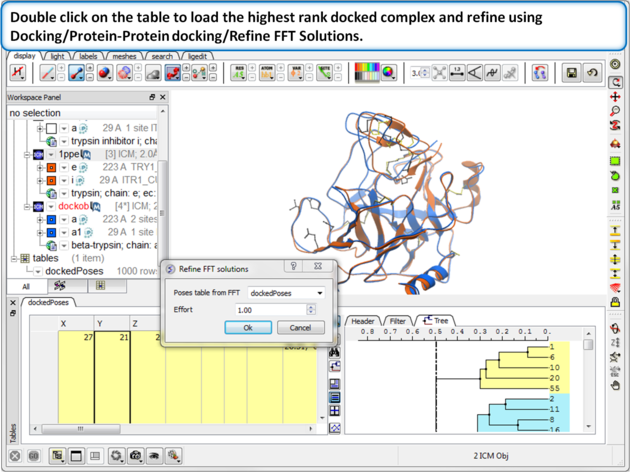 |
| Step 13: Double click on the table to load the highest rank docked complex and the new docked molecule will be loaded in the ICM workspace. Optional - refine using Docking/Protein-Protein docking/Refine FFT Solutions.
About the Results Table
|
About Refinement The refinement option is to create (if needed) a potentially more accurate atomic model of the interface rather than to pick the right solution. Therefore, for refinement select solutions from the table that look convincing (e.g. match experimental mutation observations) the clustering tree helps you do this. Delete solutions that you do not wish to refine - you can do this by making a row(s) selection then right clicking on the row header and choose delete row. When ranking it is better to consisder the lowest ener value rather than Score.