Google Search the Manual:
Keyword Search:
| Prev | ICM User's Guide 22.5 3D Ligand Editor | Next |
Workshop: Working with the ICM 3D Ligand Editor
Reference Video: Introduction to the ICM 3D Ligand Editor - Drug Design and Optimization
Case Study Overview
This workshop focuses on the optimization of first-generation EGFR (Epidermal Growth Factor Receptor) inhibitors, such as Erlotinib and Gefitinib. These compounds are highly effective against primary mutations in non-small cell lung cancer but eventually encounter resistance, specifically the T790M "gatekeeper" mutation. We will use the ICM 3D Ligand Editor to visualize these resistance mechanisms and design new modifications to improve potency and binding affinity.
Task 1: Project Initialization & Setup
|
Task 2: Visualization and Analysis
- Pocket Properties: Use the Receptor Pocket Colors dropdown to identify H-bond donors (blue), acceptors (red), and hydrophobic regions (green) [06:09].
- Surface Mapping: Toggle the Ligand Pocket Surface to identify "white space" for molecular expansion [06:29].
- Clash Detection: Enable Atomic Energy Circles. Red circles indicate steric hindrance [01:10].
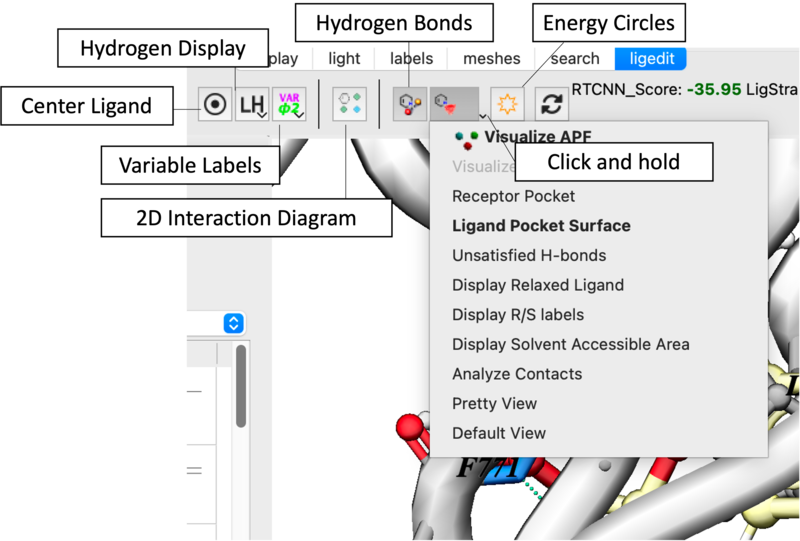
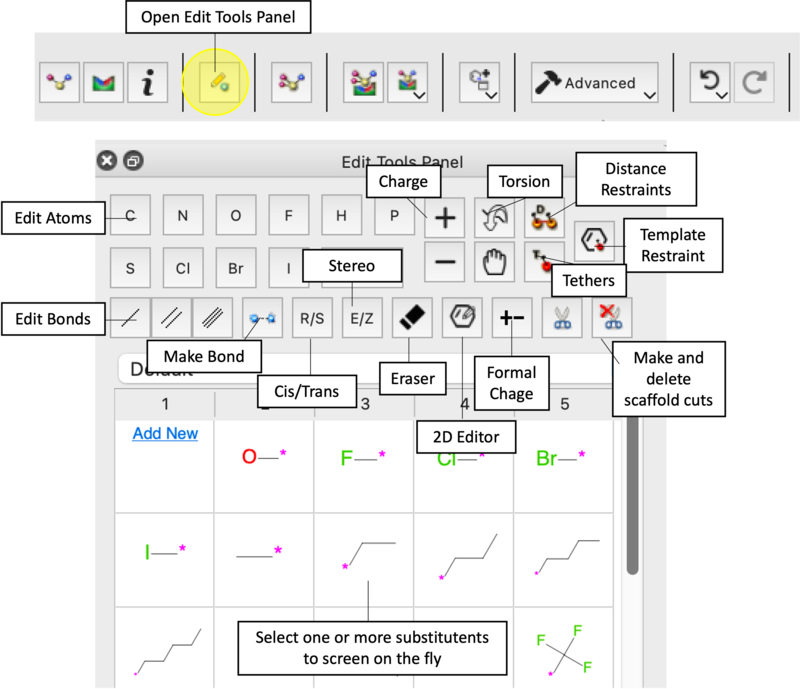
Task 3: Interactive Ligand Editing
- 2D and 3D Modifications: Open the Edit Tools Panel to modify the molecule directly in the 3D window or 2D schematic [05:55].
- Substitutions: Try replacing the amine linker with an ether or thioether [08:13]. Add a hydroxyl group to observe score improvements [09:13].
- Steric Hindrance Test: Attempt to add a bulky group near the Threonine 790 gatekeeper. Observe the red circles and Undo [09:51].
- The Eraser Tool: Use the Eraser icon to remove groups and monitor score changes [07:34].
Minimize, Refine and Redock
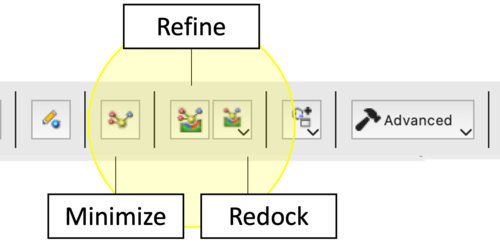
The ICM Ligand Editor utilizes three hierarchical levels of optimization to refine lead compounds. Each protocol differs in its computational cost and the degree of freedom allowed for the ligand within the receptor-defined grid potential.
| Option | Protocol | Effect | Application |
|---|---|---|---|
| 1. Minimize Ligand in Soft Grid | Local Gradient-Based Optimization. Employs the ICM force field to minimize the potential energy by adjusting internal coordinates. Uses soft van der Waals potentials to resolve steric overlaps. | Primarily optimizes torsions, bond angles, and lengths; ligand position/orientation remains essentially fixed. | Resolution of local strain or geometry distortions following manual covalent modifications or atom-type changes. |
| 2. Refine Redock using Current Pose in Soft Grid | Biased Probability Monte Carlo (BPMC) Search. A local stochastic search centered on the starting pose. It explores the immediate energy landscape to identify a deeper local minimum. | Full sampling of ligand torsions combined with small-step translations and rotations. | Optimization of binding interactions (e.g., H-bond geometries) when the general binding mode is known. |
| 3. Redock | BPMC algorithm to perform an exhaustive search of the defined grid potential. Ignores the current pose to find the global minimum. | Comprehensive sampling of the entire transitional/rotational space and all free torsions within the designated binding site. | Validation of binding modes, docking after significant scaffold hops, or when exploring potential induced-fit movements requiring a full reset of the ligand pose. |
Score and Strain

Task 4: Advanced Substituent Screening
- Position-Specific Screening: Select a hydrogen atom and choose a substituent set (Halogens, etc.) to generate a ranked Modifiers Tab [10:37].
- Large Database Screening: Right-click an atom and select Advanced - Find Group to screen ~60,000 fragments [12:19].
Task 5: Data Management and Export
- Saving Candidates: Click Add to Table for any modification with a promising score [08:38].
- Exporting Results: Export final compounds in SDF, PDB, PDF, or Excel formats [01:47].
Troubleshooting and Tips
- High Ligand Strain (>5.0): Try running another Minimization or manually adjusting the dihedral angle of the new group.
- Persistent Red Circles: This indicates a van der Waals clash. If minimization doesn't help, the group is too large for that sub-pocket.
- Score Not Updating: Click Evaluate Score manually.
- Box Size Issues: If building into a large allosteric site, you may need to increase the docking box dimensions in Setup Receptor [04:41].
| Prev Chemical Structure Analysis SAR and Library Design | Home Up | Next Ligand Docking |