[ VLS Intro | Start | Database Format | Preferences | Docking Project Table File | Run VLS in the Graphical User Interface | Parallelization | VLS Results | Post Screen Analysis | Interaction Lists | Visualization ]
| Available in the following product(s): ICM-VLS | |
| Additional Resources: Tutorial: Screen to Ricin Receptor | Tutorial: Screen to Cyclooxygenase | Tutorial: Screening a Markush Library | Command Line Docs | Video |
Virtual Ligand Screening (VLS) in ICM is performed by docking a database of ligands to a receptor structure followed by an evaluation of the docked conformation with a binding-score function. Best-scoring ligands are then stored in the multiple icm-object file. The set-up of the VLS process is largely identical to the set-up for the small molecule docking simulation (see Small Molecule Docking section).
In most cases the ligand input file will be an SDF or MOL2 file. These files need to be indexed by ICM before they can be used in VLS runs. The index is used to allow fast access to an arbitrary molecular record in a large file such as an SDF file which in some cases contains over one million compounds.
To index an sdf file:
- Click on the menu Docking/Tools/Index Mol/Mol2 File/Database to generate the index. The following data entry box will be displayed.
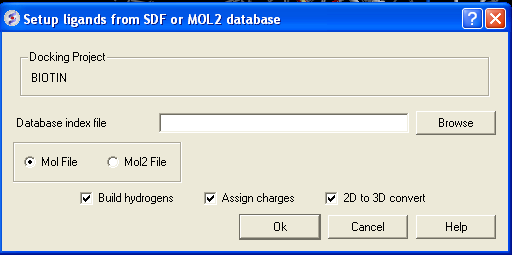
- Enter the name of your Mol/Mol2 file and enter the name you wish to call your index file.
- Select whether your file is in Mol or Mol2 format.
| NOTE: It is important to setup the VLS preferences before undertaking VLS run. Additional preferences can be changed in the docking_project_name.dtb file. The fields in this file are described here: https://molsoft.com/addons/ftp/man/dockTable.pdf |
VLS preferences can be setup by:
- Selecting the menu Docking/Preferences/Database Scan

Different options are available to select by clicking the down arrow next to the data entry field. These options are described here:
Score Threshold:
An important parameter of the VLS run is the score threshold. Docked conformations for a particular ligand will only be stored by ICM VLS procedure if its binding-score is below the threshold. The choice of the threshold can be done in two ways: based on the scores calculated by docking known ligands. Generally, a value somewhat above typical score observed for known ligands is a good guess. If no ligands are known, a pre-simulation can be run using ~1000 compounds from the target database. Using the resulting statistics for the scores, the threshold should be set to retain ~1% of the ligands.
Potential of mean force score:
Potential of mean force calculation ( pmf ) provides an independent score of the strength of ligand-receptor interaction. The pmf-parameters are stored in the icm.pmf file.
Other selection criteria:
Other selection criteria which can be changed include
Minimum/Maximum Ligand Size you wish to be screened.
Maximum number of H-bond donors
Maximum number of H-bond acceptors
Maximum number of torsions
ICM VLS uses a number of criteria to pre-select compounds before docking. Edit the project .dtb file to change their defaults. A full description of each field in the .dtb file can be found here https://molsoft.com/addons/ftp/man/dockTable.pdf
#>i DOCK1.i_maxHdonors
5
#>i DOCK1.i_maxLigSize
500
#>i DOCK1.i_maxNO
10
#>i DOCK1.i_maxTorsion
10
#>i DOCK1.i_minLigSize
100
First setup the docking project (From Set Project to Setup Batch Ligand)
To start the vls job:
- Docking/Run Docking Batch
[ Gui Jobs | Vls command line ]
If the database size exceeds several thousand compounds, it is desirable to run a number of VLS jobs in parallel to speed up the calculations. You can do this in the GUI or on the command line on a cluster or cloud.
In the GUI you can run the screening in parallel in the following ways.
- Setup the docking project.
- Check the docking preferences and the database scan preferences. If you are screening millions of chemicals it is usually a good idea to run a test screen of a smaller set so you can see the likely range of scores.
- Index the SDF file you wish to screen using Docking/Tools/Index Mol/Mol2 File/Database.
- Select the indexed database using Docking/Setup Batch Ligand/From Indexed Database.
- Docking/Run Docking VLS Batch and enter the number of CPU threads you wish to use on your local machine by entering the number in the Number of Parallel Jobs dialog window. For example if you have a 4 cpu machine with 2 threads each you could split your screening up into 8 parallel jobs.
- Then the docking will run. In your docking directory you will see multiple .ou file according to the number of parallel jobs you are runing.
Or there is a more interactive way as described below.
- Select Docking/Run Docking VLS Batch
- Select the Local VLS Batch option.
- Select the docking project name.
- Enter the compound number you would like to dock from e.g. 8001
- Enter the compound number you would like to dock to e.g. 10000
- License type: For nearly all licenses (e.g. standard ICM-VLS licenses) types you need to leave this entry blank. If you have a -vlscluster license or a molvls license (-vls) then select the option from the drop down list. Any questions first check your license.dat file or Email support@molsoft.com.
Or if you have a queuing system installed on the machine you are running the GUI you can use the tab VLS via Queueing System and this will split the jobs p via the number of jobs you specify and submit the jobs.
To run vls on the command line on a server or cloud machine.
- Setup the docking project.
- Check the docking preferences and the database scan perferences. If you are screening millions of chemicals it is usually a good idea to run a test screen of a smaller set so you can see the likely range of scores.
- Index the SDF file you wish to screen using Docking/Tools/Index Mol/Mol2 File/Database.
- Select the indexed database using Docking/Setup Batch Ligand/From Indexed Database. Check that the path to the database is correct in the docking_project.dtb file - you might need to add the path in the fields shown below.
#>s dockCOX.s_dbFile
/home/molsoft/celebrex50.sdf
#>s dockCOX.s_dbIndex
/home/molsoft/celebrex50.inx
- Send the jobs using _dockScan.
The easiest way to view the results of a VLS run is to make a hitlist. This was described earlier in the hitlist section of the small molecule docking chapter of the manual. Other ways of manipulating VLS data are described here:
[ Sort Hitlist | Plot | Complex & H-bond Display | Calculate Properties | APF Cluster | Interaction Fingerprints ]
Compounds can be sorted according to their SCORE and other properties in the hitlist. To sort the table single click on the column header - see the tables section of this manual for more information about manipulating tables.
The hitlist generated from docking or screening typically contains many columns with numerical data, such as Score, RTCNN score, MolWt, LogP, Number of H-bond donors/acceptors, and others. Visualizing relationships between these columns can be a powerful way to explore trends and prioritize compounds. For example you may want to plot Score (Physics-Based Score) vs RTCNN Score and select those with low values for each. See the data plotting section of the manual here.
When you make the hitlist you will see a panel next to the chemical table called Tools. The options here can be used to:
- Check the option display Hbonds if you would like to display hydrogen bonds when you browse the hitlist.
- Check the option make complex structure if you would like the ligand-receptor complex to be displayed when you browse the hitlist.
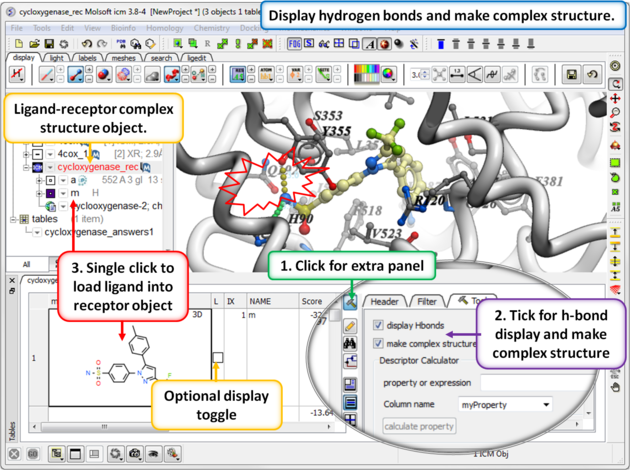
The hitlist tools allow you to calculate a range of property expressions that provide deeper insight into how all the ligands in your hitlist interact with the receptor. By pressing the 'gear' button in the top-right corner of the hitlist table, you can open the Tools panel and access these calculations through the Property Expression menu.
Options include measuring ligand charge, buried surface area, conformational volume change, hydrogen bond interactions, pharmacophore RMSD, cis/trans amide angle, and spatial relationships to receptor selections. Additional functions include pose similarity, interaction fingerprint clustering, and automatic listing of residue-level interactions. These properties help assess ligand fit, stability, and interaction quality within the binding site, supporting more informed analysis and decision making when choosing chemicals for experimental testing.

The options include:
-
Calculate charge of the ligands in the hitlist.
Computes the net molecular charge of each ligand. Charge influences solubility, membrane permeability, and electrostatic interactions with the binding site.
-
Calculate Buried Surface Area (BSA) of the ligands in the hitlist.
Measures how much ligand surface becomes inaccessible to solvent upon binding. Higher BSA often correlates with stronger binding because more of the ligand contacts the protein surface.
-
Calculate volume change of the ligands in the hitlist.
Compares the bound conformation to a low-energy free conformation. Large volume change can indicate distortion to fit the pocket, which may reduce affinity due to strain or entropy. Small change suggests a good fit with minimal adjustment.
-
Calculate whether the ligand makes a hydrogen bond with a selection.
Select a receptor residue or atom first. The scoring function ranges from -2 to 0; more negative values indicate stronger hydrogen bonds. The score uses donor-acceptor distance and angle to approximate typical hydrogen bond energetics. A value near 0 means no significant interaction; values approaching -2 reflect optimal geometry.
-
Calculate pharmacophore RMSD.
Measures the root-mean-square deviation between a ligand’s pharmacophoric features and a reference model. Requires an object named pharma with the pharmacophore loaded in the workspace. Useful for assessing alignment to a pharmacophore hypothesis.
-
Calculate cis/trans amide angle of the ligands in the hitlist.
Checks amide bond geometry. Most amides are trans; cis can be less stable or less favorable for binding. Helps flag unusual or strained conformations.
-
Calculate distance to selection.
Select a receptor atom or residue first. Computes the distance between the selection and each ligand, useful for measuring proximity to key residues or functional groups.
-
Calculate non-overlap with selection.
Select a receptor residue first. Evaluates how much of the ligand avoids steric overlap with the selection to help detect potential clashes.
-
APF pose similarity to a selected ligand.
Uses Atomic Property Fields (APF) to compare ligand poses against a chosen reference ligand, incorporating shape and pharmacophoric properties to identify pose mimicry.
-
Calculate and cluster the interaction fingerprints.
Encodes ligand-receptor interactions into a binary fingerprint (e.g., hydrogen bond donor/acceptor, apolar). The resulting column can be clustered to group ligands with similar interaction patterns.
-
Calculate a list of the interactions.
Generates columns per pocket residue with 1/0 values indicating whether the ligand forms donor, acceptor, or apolar interactions. Produces a residue-level interaction map for the hitlist.
Each of these properties will be placed in the hitlist table as a new column. You can choose a column name before calculating using the Column name dialog box. Click on the Calculate property button to run the calculation.
- Make the hitlist (Docking/Make Hitlist)
- Press the Cluster by APF button on the Tools panel (right hand side of hitlist table).
- A tree will be displayed clustering the pose similarity of the compounds by APF fit.
- The distance matrix is added to the hitlist.
An interaction fingerprint can help choose docked ligands with common interactions or a diverse set of interactions.The fingerprint is a bit string representing a contact with each atom. For specificity there is a special subset of bit strings which represent hydrogen bonding. An alternative to interaction fingerprints is to cluster by 3D pharmacophoric similarity using APF.
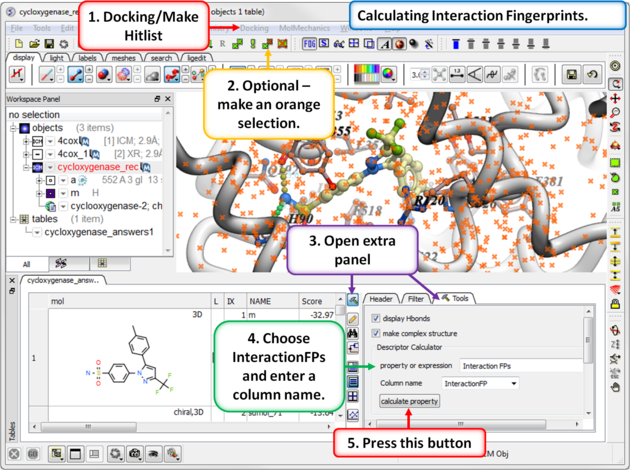 |
Step 1: Calculate Interaction Fingerprints
|
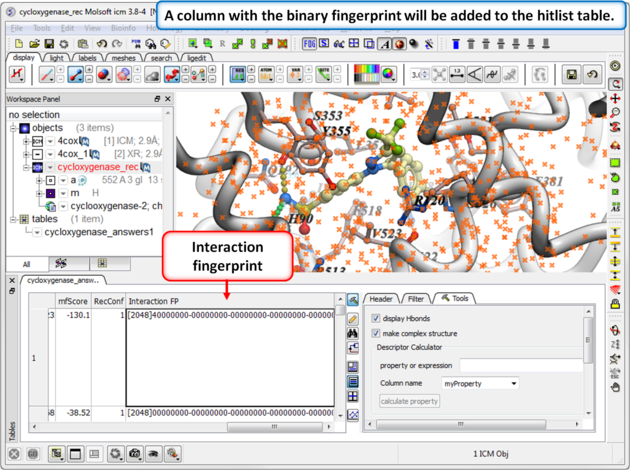 |
| Step 2: Once the calculation has finished a column will be added to the hitlist table. |
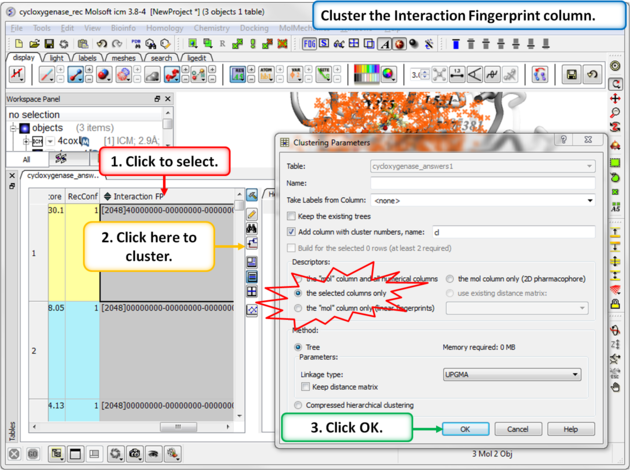 |
Step 3: Cluster the Fingerprints
|
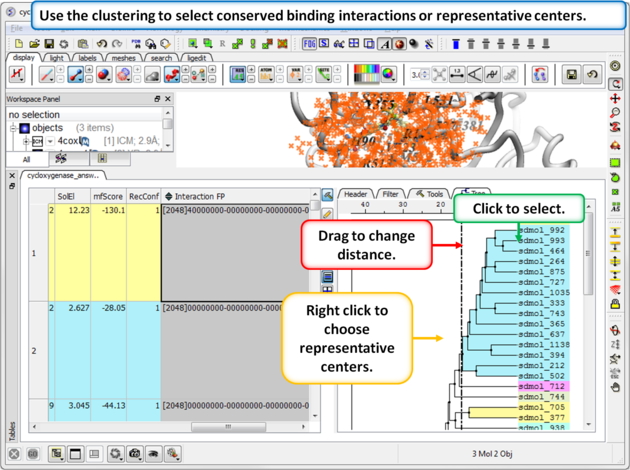 |
Step 4: Select representative centers or conserved binding interactions.
|
In these lists, residues are labeled (1 or 0) according to their interaction type or property, helping you interpret their role in ligand binding:
- apo - Apolar. The residue interacts with the ligand through hydrophobic (non-polar) contacts, such as van der Waals interactions. These are important for stabilizing ligand binding through shape complementarity and desolvation.
- hba - Hydrogen bond acceptor. This residue accepts a hydrogen bond from a donor group on the ligand.
- hbd - Hydrogen bond donor. This residue donates a hydrogen bond to an acceptor group on the ligand.
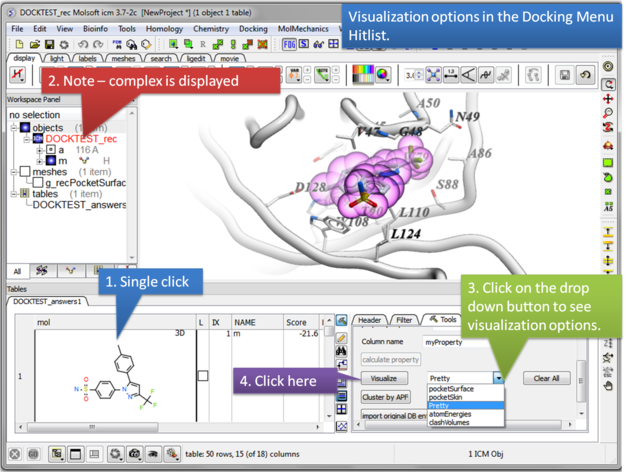
Once you have made a hitlist of the screening results you can use the panel on the right hand side of the hitlist to change visualization options. The available options are:
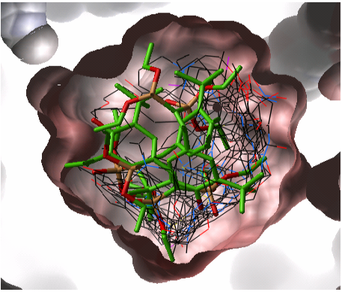
- pocketSurface = The surface of the ligand binding pocket. White=aromatic lipophilic, Green=non-aromatic other (mostly aliphatic) lipophilic surface, Red=hydrogen bonding acceptor potential, Blue=hydrogen bond donor potential
- pocketSkin = A skin representation of the neighboring residues.
- Pretty = A default representation for ligand and receptor (see image below).
- atomEnergies = An energy value for each atom in the ligand is displayed.Green and low numbers indicate a positive contribution to binding.
- clashVolumes = Highlights any clashes between ligand and receptor.