| Tutorial |
| Available in the following product(s): ICM-Pro |
This method computes the change in protein stability upon mutation of a single residue.
Getting Started: A PDB structure or ICM object containing the protein complex is needed. A graphical selection of the residue to be mutated is then made, the mutant amino acid (e.g., "ala" or "all" for calculation of the energy for all natural amino acids) is then selected.. "Scan Sequence" allows you to mutate all the residues in the sequence to the specified amino acid.
The free energy change in protein stability is computed as follows:
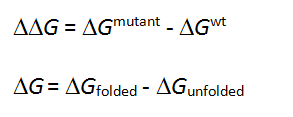
CalculationThe free energy of the unfolded and misfolded states is approximated by a sum of the residue-specific energies. The residue-specific energies were derived empirically using a large set of experimental data. Mutation of a given residue is followed by Monte Carlo simulations with flexible side chains for the mutated residue and its neighboring residues. The rest of the protein structure is considered rigid. A positive energy value indicates that the mutation is likely to be destabilizing.
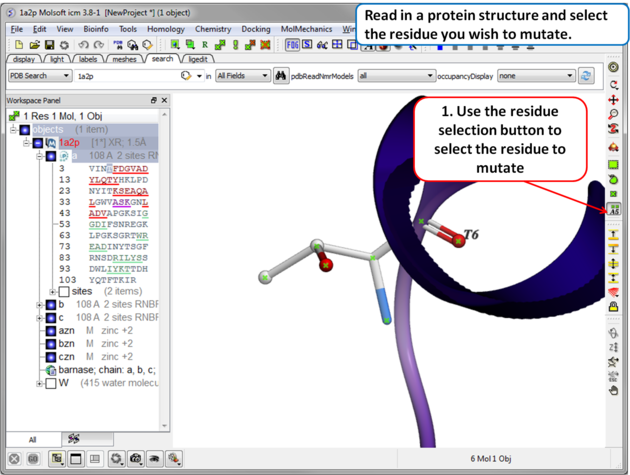 |
| Step 1 Read in the PDB file of interest and select the residue you wish to mutate. |
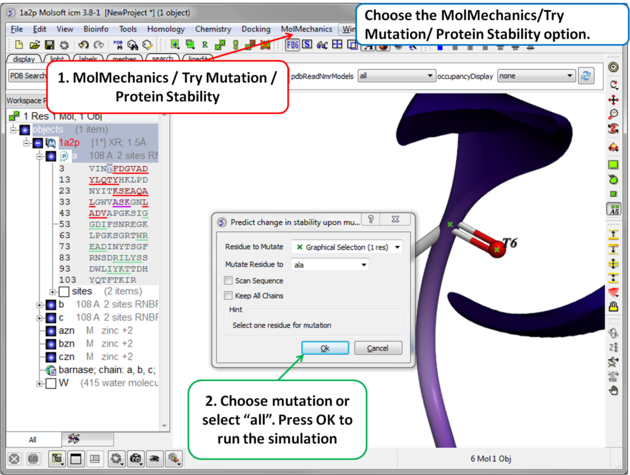 |
| Step 2 Choose the option MolMechanics/Try Mutation/ Protein Stability and a dialog box as shown above will be displayed. If you have selected a residue it should be reported in the "Residue to Mutate" box. Select a residue to mutate to from the drop down list or select "all" to try all 20 natural amino acids. The "Scan Sequence" option allows you to mutate all the residues in the sequence to the specified amino acid. The "Keep All Chains" keeps all the chains in the mutated structure, otherwise, all chains except the one containing the mutatable residue will be deleted". |
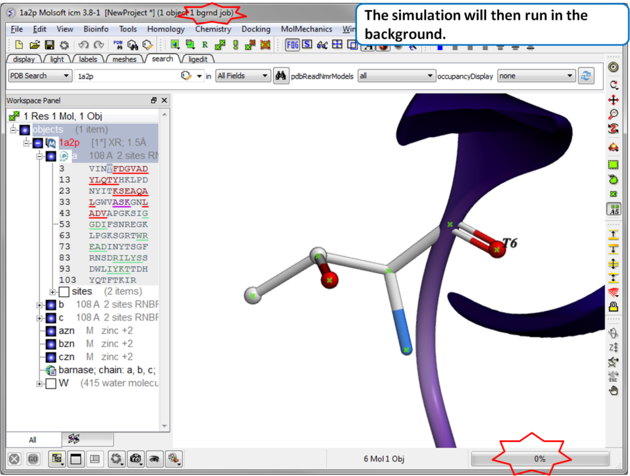 |
| Step 3 The simulation will run in the background and may take a while to finish if the option "all" or "Scan Sequence" is selected.
|
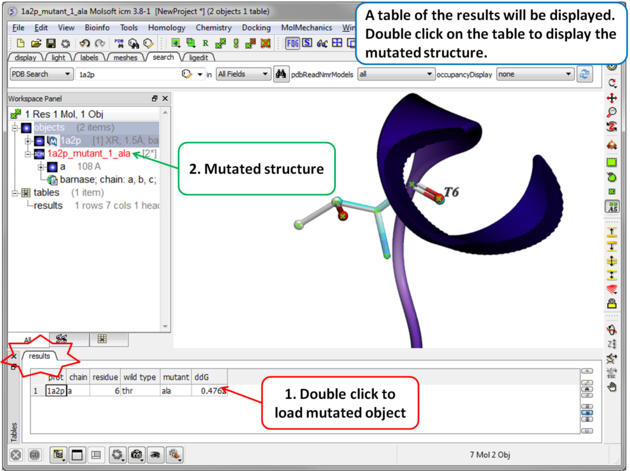 |
| Step 4 A table reporting the free energy change in protein stability will be displayed (ddG kcal/mol). Double click a row of the table to load and display the mutated structure. |
- ddG > 0: Indicates the mutant is less stable than the wild-type protein. The mutation increases the free energy of folding, making the folded state less favorable.
- ddG < 0: Indicates the mutant is more stable than the wild type, as the mutation decreases the free energy of folding, making the folded state more favorable.
- ddG ≈ 0: Suggests the mutation has little to no effect on stability.