[ GPCR Model | Loop model tutorial | Mutation Binding | Mutation Stability ]
Overview
This tutorial will take you through the basics of protein modeling. Topics include:
- GPCR modeling example
- Loop Modeling
- Predicting the Effect of Mutation on Binding
- Predicting the Effect of Mutation on Protein Stability
Background
ICM has an excellent record in building accurate models by homology. The ICM modeling procedure builds the framework, shakes up the side-chains and loops by global energy optimization. You can also color the model by local reliability to identify the potentially incorrect regions of the model. ICM also offers a fast and completely automated method to build a model by homology and extract the best fitting loops from a database of all known loops. It just takes a few seconds to build a complete model by homology with loops. Some selected publications related to modeling and structure determination are listed here.
Abagyan, R.A., and Totrov, M.M. (1994). Biased Probability Monte Carlo Conformational Searches and Electrostatic Calculations for Peptides and Proteins. J. Mol. Biol., 235, 983-1002
Cardozo, T., Totrov, M., and Abagyan, R. (1995). Homology modeling by the ICM method. Proteins: Structure, Function, Genetics, 23, 403-414
Abagyan, R., and Totrov, M. (1999). Ab initio folding of peptides by the optimal-bias Monte Carlo minimization procedure. Journal of Computational Physics, 151, 402-421
Maiorov, V.N., and Abagyan, R.A. (1997). A new method for modeling large-scale rearrangements of protein domains. Proteins, 27, 410-424
Schapira, M., Totrov, M. and Abagyan, R. (2002). Structural Model of Nicotinic Acetylcholine Receptor Isotypes Bound to cetylcholine and Nicotine. BMC Structural Biology 2:1
In this example we will build a homology model of GPR120 which is a member of the Class A GPCRs. GPR120 is gaining particular interest because it has been shown to be involved in the insulin-sensitizing effects of omega 3 fatty acids as well as inflammation. GPR120 dysfunction is responsible for reduced fat metabolism, thereby leading to obesity.
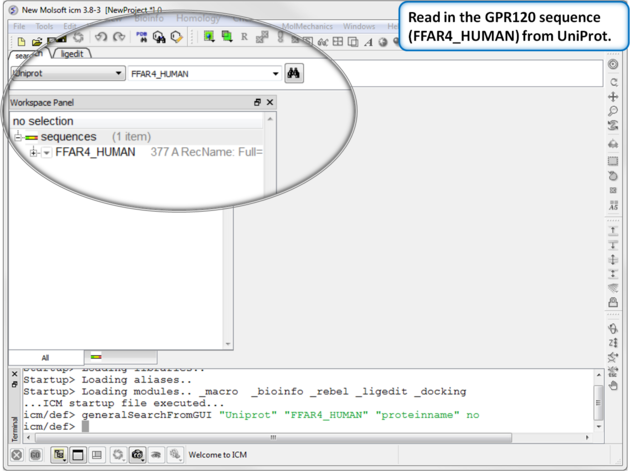 |
| Step 1: Read in the GPR120 sequence (FFAR4_HUMAN) from UniProt. |
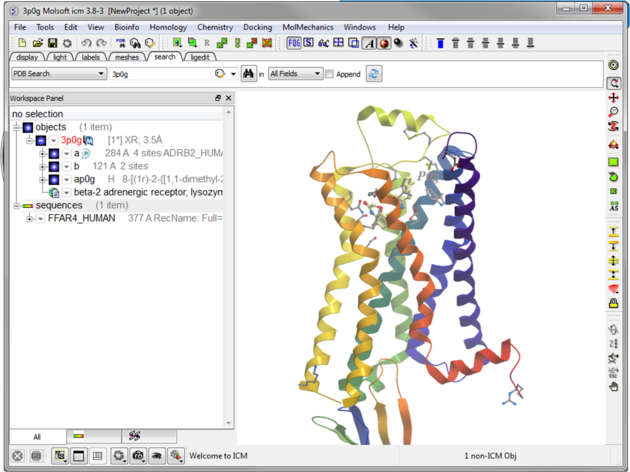 |
| Step 2: Read in the template structure 3P0G. |
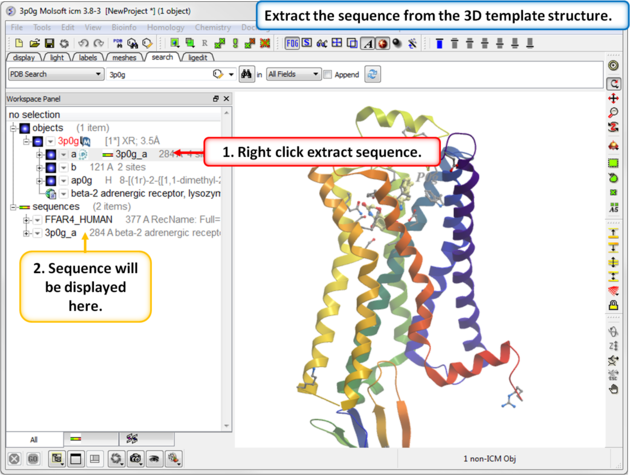 |
| Step 3: Extract the sequence from the 3D template structure. |
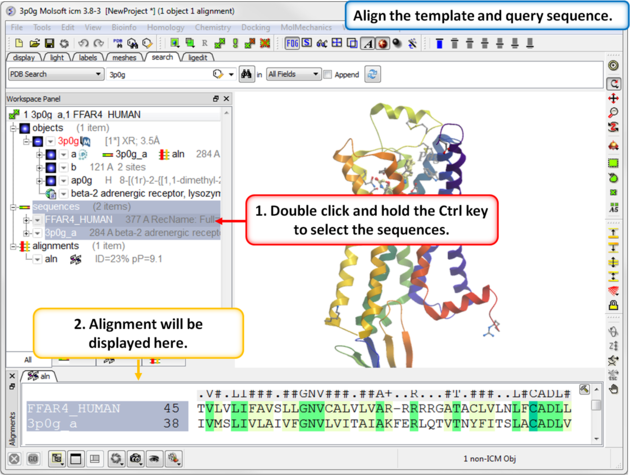 |
| Step 4: Align the template and query sequence. |
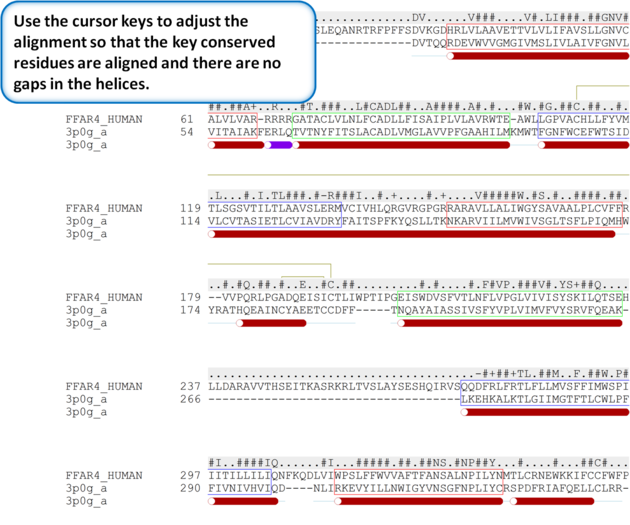 |
| Step 5: Use the cursor keys to adjust the alignment so that the key conserved residues are aligned and there are no gaps in the helices. |
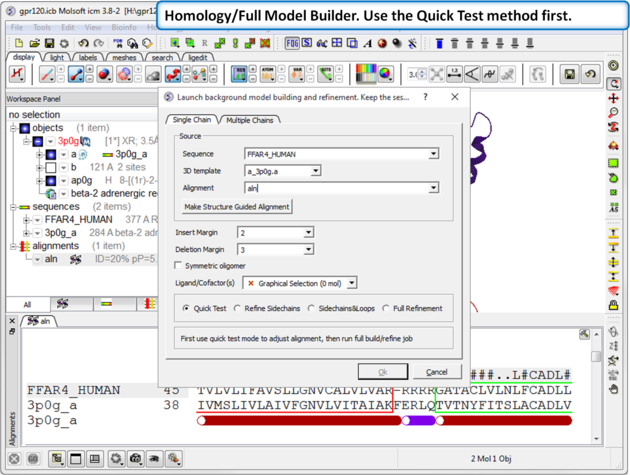 |
| Step 6: Choose the option Homology/Full Model Builder. Use the Quick Test method first to inspect the quality of the model-sequence alignment. |
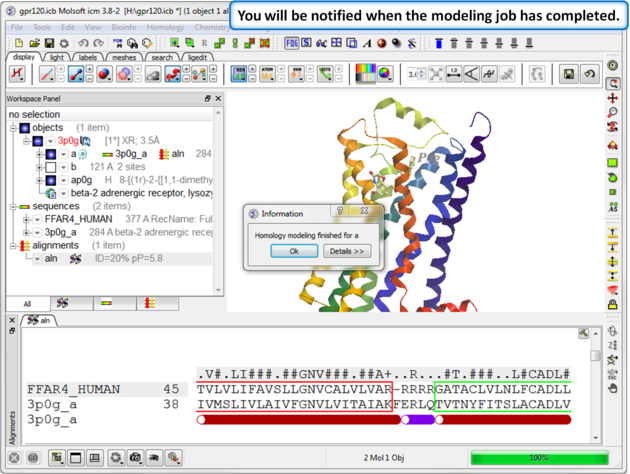 |
| Step 1: A dialog box will be displayed when the modeling job has finished. |
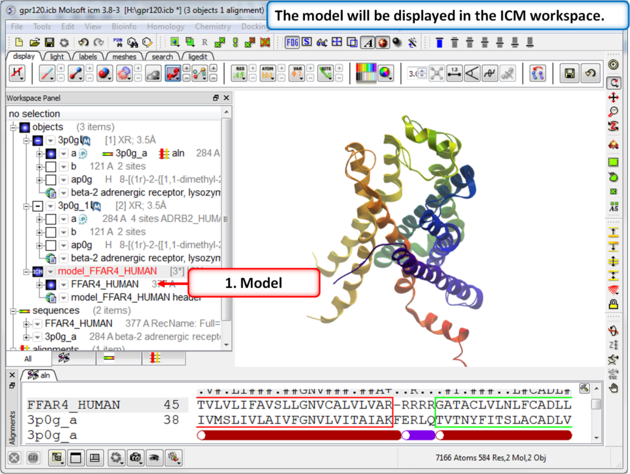 |
| Step 8: The model will be displayed in the ICM Workspace. |
In this example we will use the high precision loop modeling method Arnautova et al Proteins. 2010, 79:477 to model a loop region in Anthrax Protective Antigen which contains a proline residue where the cis/trans conformation is not clear from the available X-ray crystal structures. We will determine the most energetically favorable conformation of the proline and the neighboring loop residues.
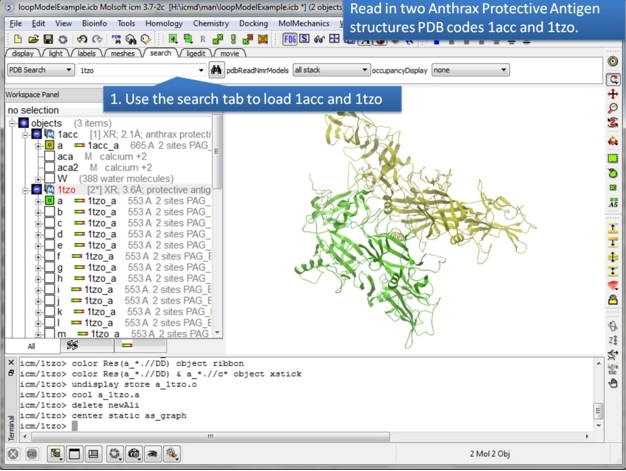 |
| Step 1: Use the search tab to read in two pdb structures 1acc and 1tzo. |
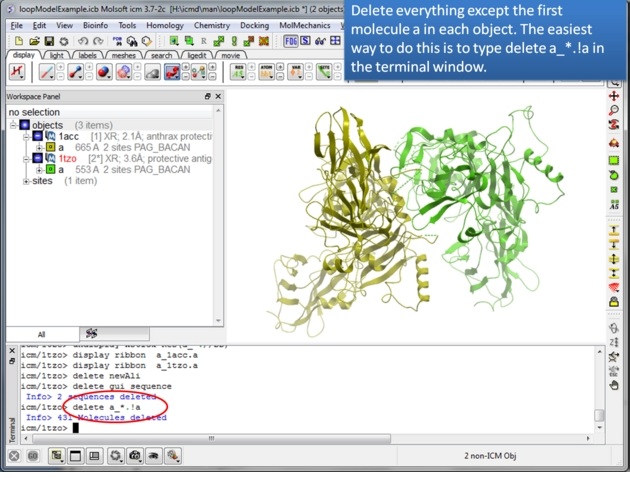 |
| Step 2: Delete everything except for the first molecule of each protein. You can use the command line as suggested to delete the other molecules or you can: -use the CTRL button and click on them - right click and select delete. |
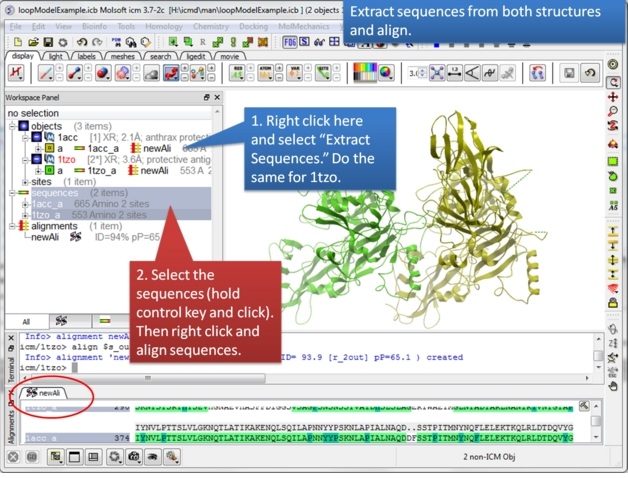 |
| Step 3: Extract hte sequences from both receptor by right clicking on the molecule in the ICM workspace. The alignment makes it easier to superimpose the loop and select the loop in both structures. |
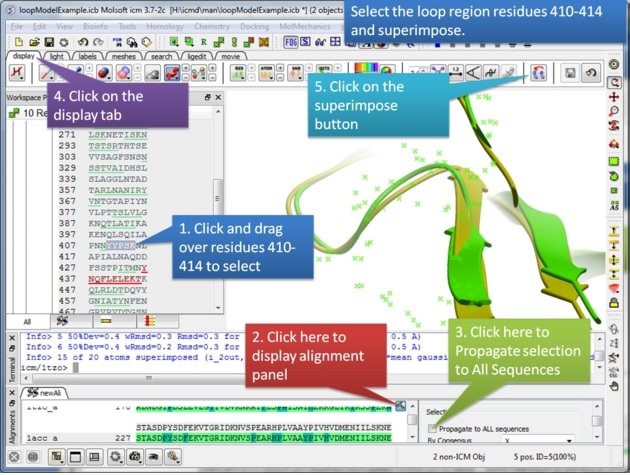 |
| Step 4: Select the loop region from residues 410-414. |
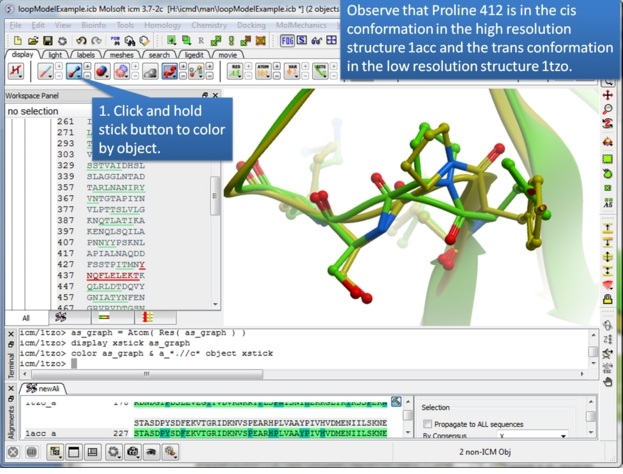 |
| Step 5: Take this opportunity to look at both structures and observe that one is in the trans conformation and the other in the cis. |
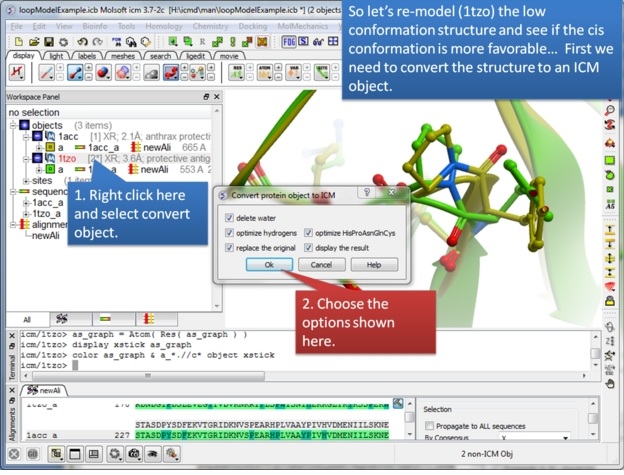 |
| Step 6: We will re-model the lower resolution structure and so we need to convert it into an ICM object. |
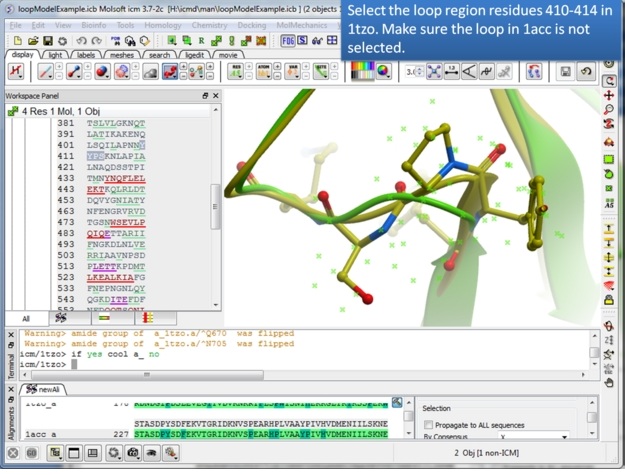 |
| Step 7: Select the loop region in 1tzo only - do not select the loop in both proteins. |
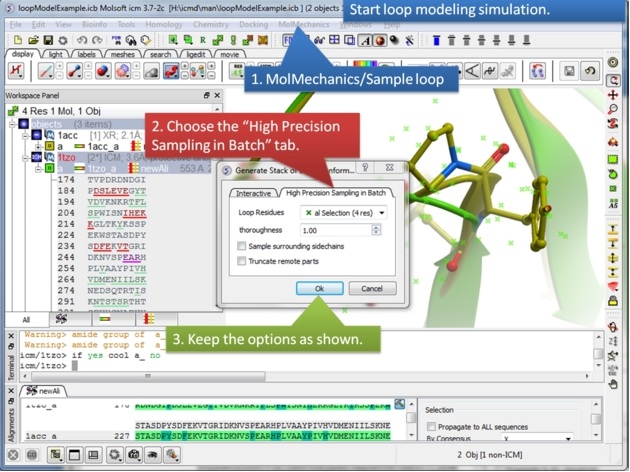 |
| Step 8: Start the loop modeling using the option in the MolMechanics menu. |
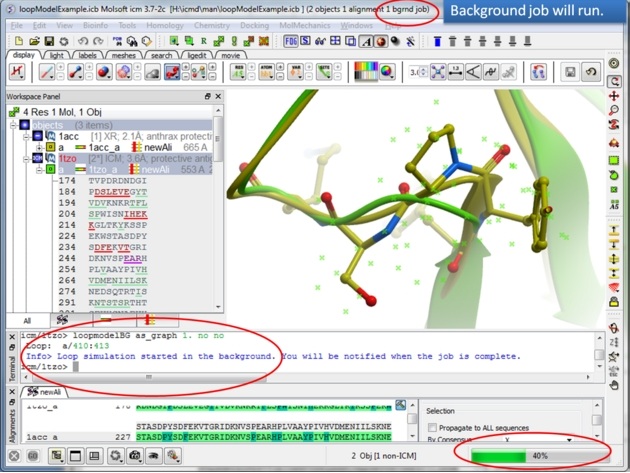 |
| Step 9: The docking will run in the background. |
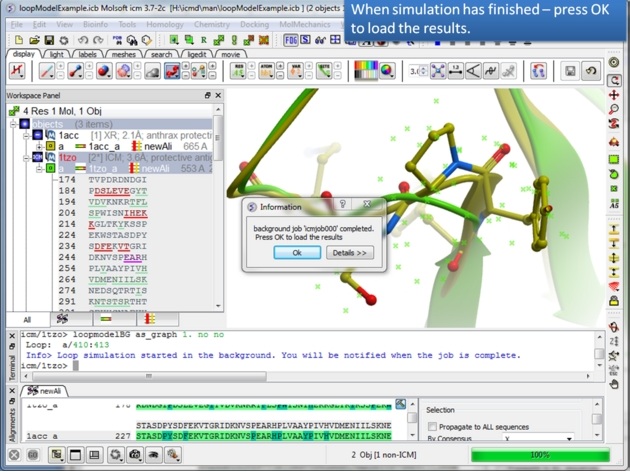 |
| Step 10: You will be notified when the simulation is finished. |
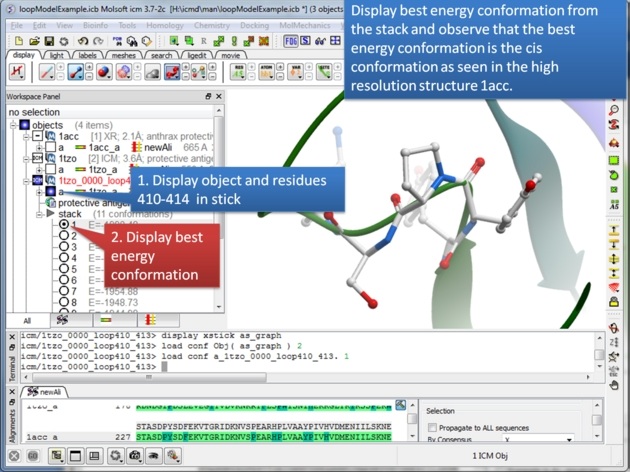 |
| Step 11: The best energy conformation of the proline in the loop is the cis conformation. |
The effect of mutation on protein binding can be predicted.
In this example the effect of mutation on binding between chymotrypsin and an inhibitor is predicted (using PDB 1CBW). LYS15 in the inhibitor makes a key binding interaction with the protease and mutation to alanine leads to a significant drop in the binding free energy.
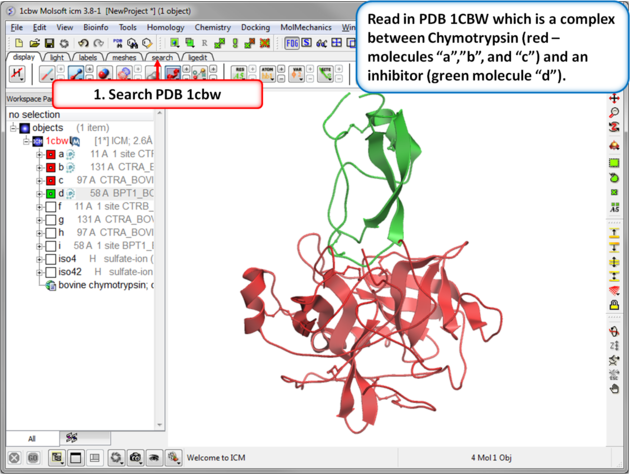 |
| Step 1: Read in the Chymotrypsin-Inbibitor complex (PDB: 1CBW). Notice that the inhibitor (green) interacts with three molecules of chymotrypsin (red). |
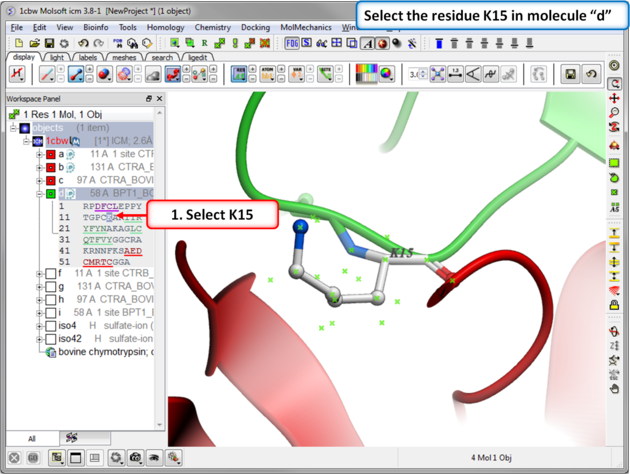 |
| Step 2: Select the residue to mutate. Select residue K15 in the inhibitor - molecule "d". |
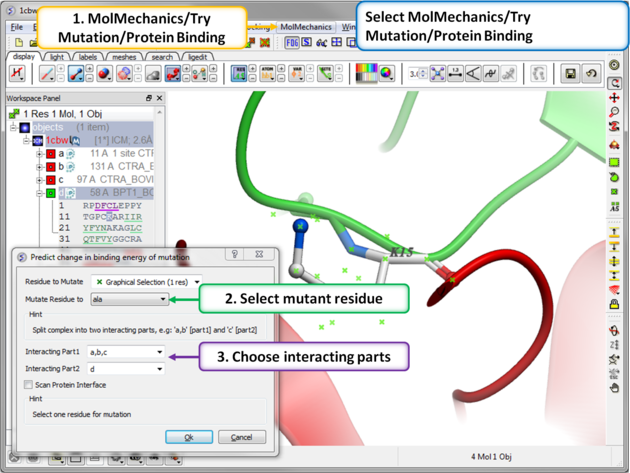 |
Step 3: MolMechanics/Try Mutation/Protein Binding.
|
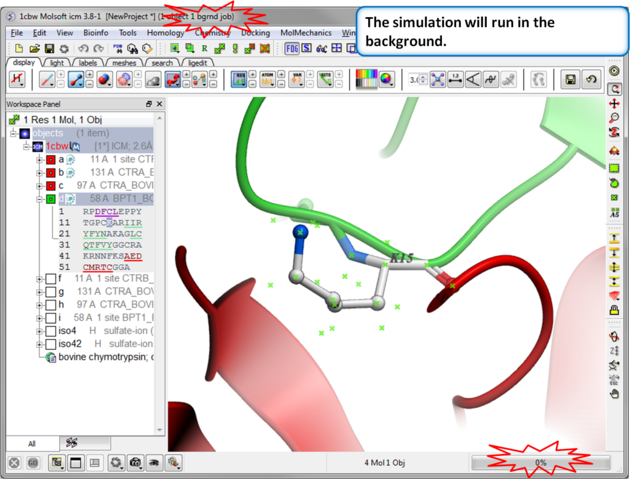 |
| Step 4: Prediction Running. Prediction will run in the background. |
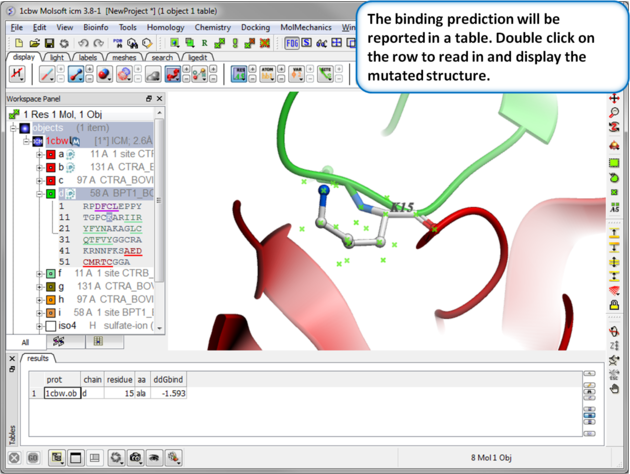 |
| Step 5: Results. A table reporting the binding free energy in kcal/mol (ddGbind) will be displayed. Double click a row of the table to load and display the mutated structure. |
The effect of mutation on protein stability can be predicted. In this example we mutate residue F26 to Ala in the crystal structure of bovine Acyl-CoA binding protein. The experimentally calculated value of this mutation is 1.54 kcal/mol ( Kragelund et al. NSB 6, 594:601 (1999) ) .
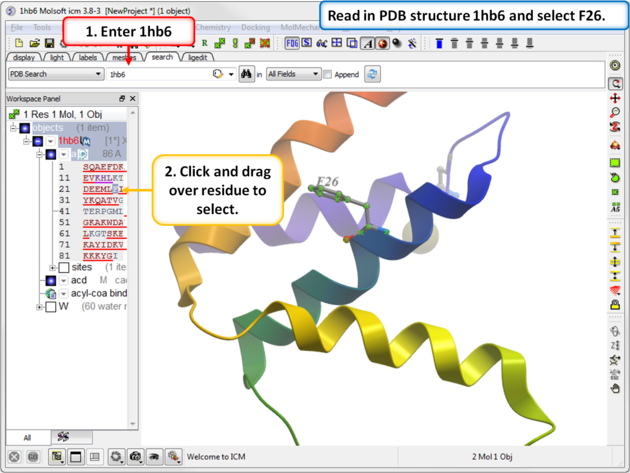 |
| Step 1: Read in PDB 1hb6 and select residue F26. |
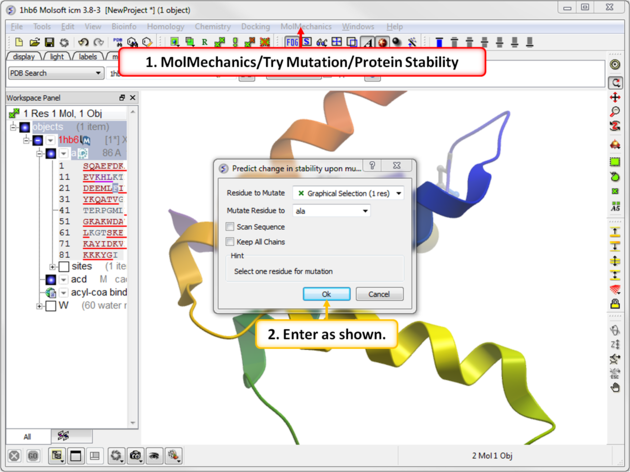 |
| Step 2: Choose the menu option MolMechanics/Try Mutation/Protein Stability and choose Ala mutation. |
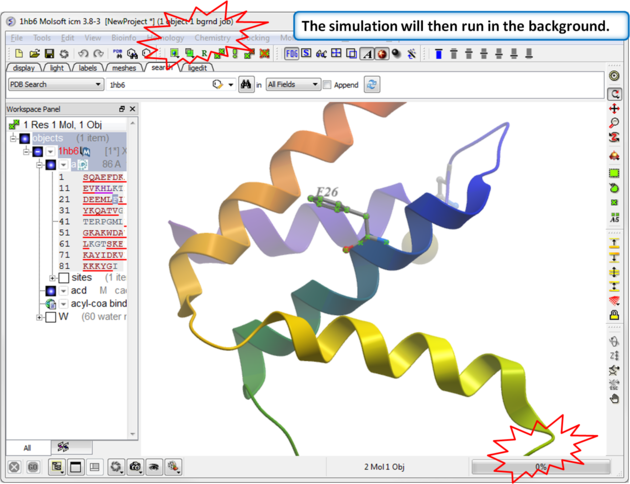 |
| Step 3: The simulation will run in the background. |
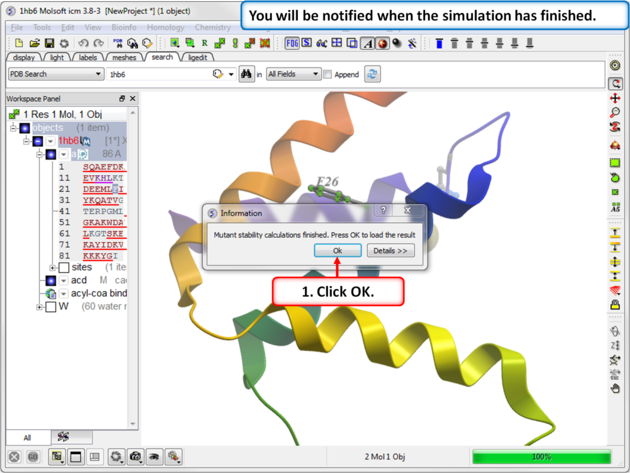 |
| Step 4: You will be notified when the simulation is complete. |
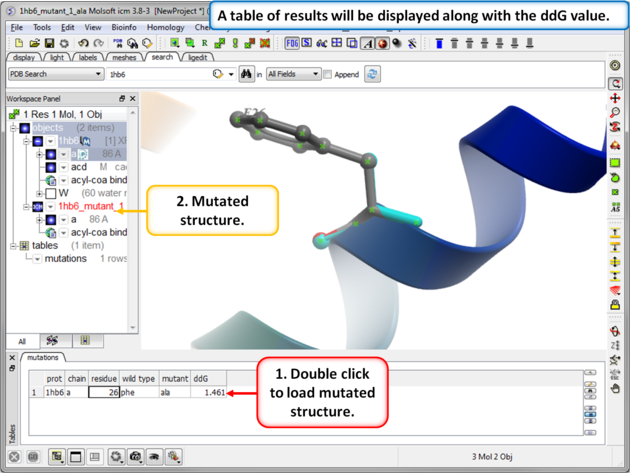 |
| Step 5: The predicted ddG value should be close to the experimental value of 1.54kcal/mol. Double click on the results table to load and view the mutated structure. |