[ defCell | accFunction | gapFunction | I_out | M_out | R_out | R_2out | S_out | S_proteinTags | swissFields | readMolNames | Selection variable | as_out | as2_out | vs_out ]
Resolution
and
MaxHKL
functions if cell parameters are not provided as arguments.
Default: {1. 1. 1. 90. 90. 90.}
|
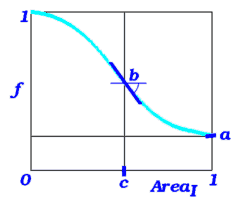
the real array of the solvent accessibility penalty parameters (as described in
Batalov and Abagyan, 1999).
It contains the values of a, b, c and E damping parameters for amino acid substitution scores. Generally, if a residue is completely buried ( Area=0), its substitution scores will be used without changes. If it is completely exposed, its substitution scores will be multiplied by the minimal possible value of a. Between these cases the substitution scores are modulated by a smooth ("arctangent") function with a saddle point at Area=c, where the slope will be -b. The fourth parameter is reserved for development. |
This definition is effectively implemented in the
Align( seq_1 seq_2 area ) },
Score functions and find database command.
Default: {0.33 2.35 0.211, -15.0}.
See also:
alignMethod .
Batalov and Abagyan, 1999).
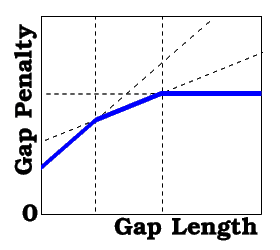
ATTENTION: at the present time this
gapFunction is only active when
alignMethod =2.
The first two values replace
gapOpen and
gapExtension traditional values.
If present, the third element of the array represents the length of the gap,
starting at which further gapExtensions become equal to the
fourth element of the array. Likewise, if more elements are present, they
represent pairs of the threshold lengths of the gap and the new gapExtensions
values. For example,
gapFunction = {2.4 0.15 10. 0.05 20. 0.}- gap penalty=2.25+0.15*L for L={ 0..10} (and for L=1 it is 2.4=
gapOpen), - gap penalty=3.25+0.05*L for L={11..20} and
- gap penalty=4.25 for L>20
|
The calculations are fastest for the traditional two-element gapFunction.
The three- or four-element gapFunction invokes the optimized routines and
is 50-70% slower. The general kind gapFunction costs approximately 70-90%
additional time for every pair of gapFunction values.
If the last gapExtension is zero, it may be omitted.
This definition is effectively implemented in the
Align , Score functions and
find database search command.
|
Default: {2.4 0.15}.
Recommended (put it in your
_startup file): gapFunction = {2.4 0.15 10.}
This set will produce fast and structure-like alignments.
See also
alignMethod, and accFunction (the accessibility attenuation parameters).
matrix
in which the output of some commands is stored.
Functions returning in R_out:
Axis# middle point of the axisDisgeo# returns error sum of negative scaled eigen values in R[0], and first three 3 scaled eigen valuesLinearFit# residualsXyz(.. ) returns inverse transformation | learnreturns model accuracy and stds.
rarray. Used in addition to R_out.
sarray of sequence patterns that can be used for detection or deleting those tags from a sequence using Trim ( seq S_tags ) function.
If the field name starts from a minus ('-'), this field will be ignored in
the feature table list. Also note that variable i_2out (after read sequence) contains the beginning of mature sequence.
Example:
swissFields={"-HELIX ","-COIL ","-STRAND","-TURN "}
# to suppress the FT records with the secondary structure info
...
M END
> <CAT_NO>
R150002
> <NAME>
(5-OXO-HEXAHYDRO-PYRANO[3,2-B]PYRROL-1-YL)-ACETIC ACID METHYL ESTER
$$$$readMolNames = {"<CAT_NO>" "<NAME>"} # useful for Sigma-Aldrich filesAnother example:
readMolNames = {"<CODE>" "<IUPAC_NAME>"} # useful for ACD databaseExamples:
cc = a_//ca # created named selection variable cc
show cc & a_/3:15 # use it in the expressionExample:
cc = "a_//ca" # in this case cc is a string, not a selection
show $cc & a_/3:15 # $cc is replaced by a_//ca before parsing
How to store and exchange selections in strings:Examples of using the String ( os|ms|rs [name|number] ) function to return a residue selection:
l_showResCodeInSelection = yes # the default
res_str = String( Res(Sphere( a_H [1] a_A//!h*,ca,c,n,o 3.5 ))) # same as with option String(.. name)
show res_str
a_2c0cb.b/^T159,^S205,^K209,^Y224,^V248,^Y275,^L305,^M356,^N361
# or
l_showResCodeInSelection = no
res_str = String( Res(Sphere( a_H [1] a_A//!h*,ca,c,n,o 3.5 ))) # same as with option String(.. number)
show res_str
a_2c0cb.b/159,205,209,224,248,275,305,356,361selection variable where
some commands or functions store their output:
If atoms a_1./3/ca,c,n relate to atoms a_2./45/ca,c,n, then the first
set will end up in as_out and the second in as2_out.
selection ) returned by the following commands and functions:
See also: as_out.
- read variable saves a selection of loaded variables;