[ Setup ]
The key docking steps are:
- Setup the Docking Project.
- Check Docking Preferences.
- Choose Ligand or Database and Start Docking Simulation
- View Docking Results
[ Adjust Binding Site - Optional | Check Maps - Optional | Docking Preferences - Optional ]
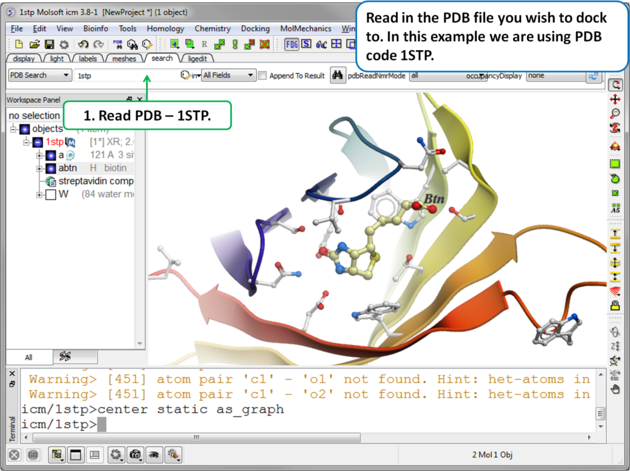 |
| Step 1. Read in the PDB file. In this example we will re-dock the biotin to streptavidin using PDB code 1STP. |
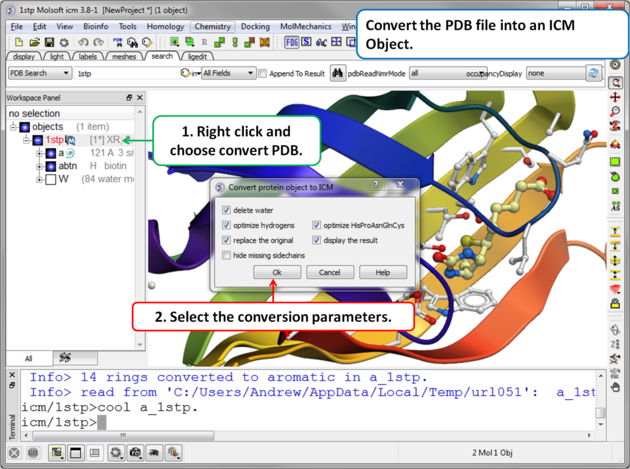 |
Step 2. Convert PDB
|
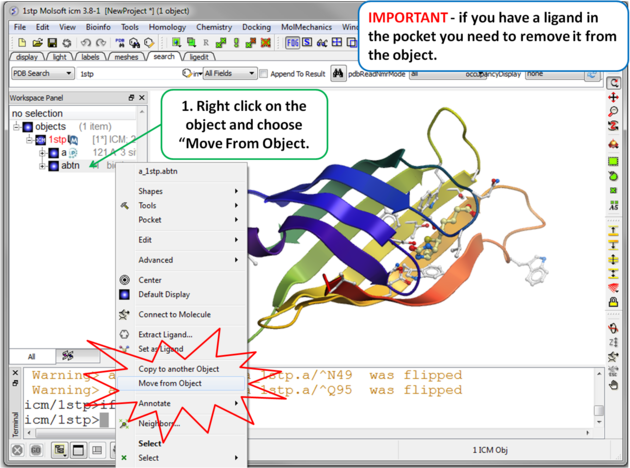 |
| Step 3 Move the Ligand out of the Pocket IMPORTANT! If you are redocking a ligand please remember to remove the ligand from the ligand binding pocket otherwise the ligand will be included in the docking maps and you will not be able to re-dock it correctly. To remove a ligand from an object - right click on the ligand in the ICM Workspace and select "move from object". Simply undisplaying the ligand is NOT sufficient. |
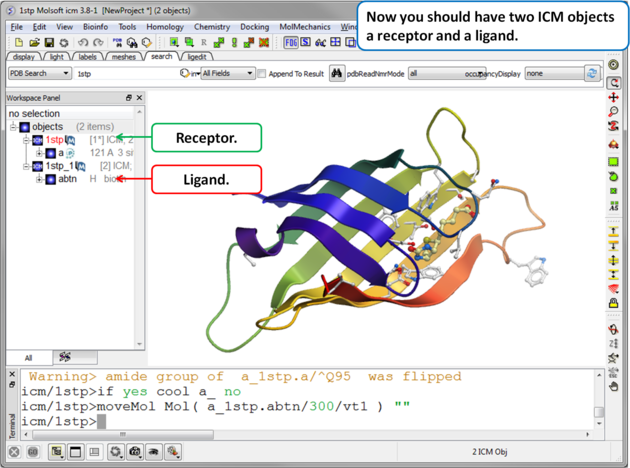 |
| Step 4. Check the Ligand. You should have two objects in the ICM Workspace - the receptor (e.g. 1stp) and the ligand (1stp_1). For clarity you can rename them by right clicking on the name in the ICM workspace and choose rename. This is a good opportunity to double check that the bond types and formal charge of the ligand are correct. You can edit them as described here. |
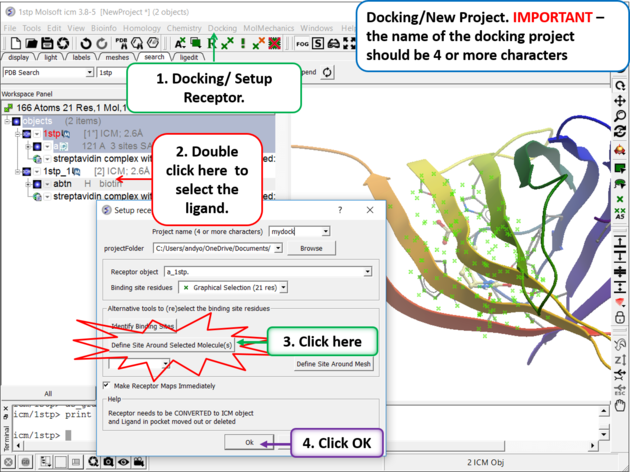 | |
Step 5. Docking/Setup Receptor
|
- Choose the option Make receptor maps immediately. You can uncheck this if you are going to do 4D or covalent docking because the maps are re-made with these options.
- Enhanced Born Scoring option is available. Enhanced Born Scoring precalculates Born radii using a Poisson-Boltzmann calculation based on a set of baseline radii. Born radii reflect how exposed each atom is to solvent, and they are used in implicit solvation models to estimate electrostatic solvation energy. The Poisson-Boltzmann equation provides a more accurate way to calculate electrostatics in a solvated environment, and this method refines initial (baseline) estimates of atomic exposure to improve accuracy. This can be especially helpful in electrostatic calculations for highly polar or deeply buried binding pockets. The main time cost occurs during the generation of the energy maps and setting up the receptor, but this does not affect the actual docking or virtual screening time.
- Click on the OK button.
| NOTE: At this stage of the docking setup it is a good idea to keep an eye on the terminal window. Instructions and any error messages will be displayed in the terminal window. If you do not see the terminal window select Windows/Terminal Window. |
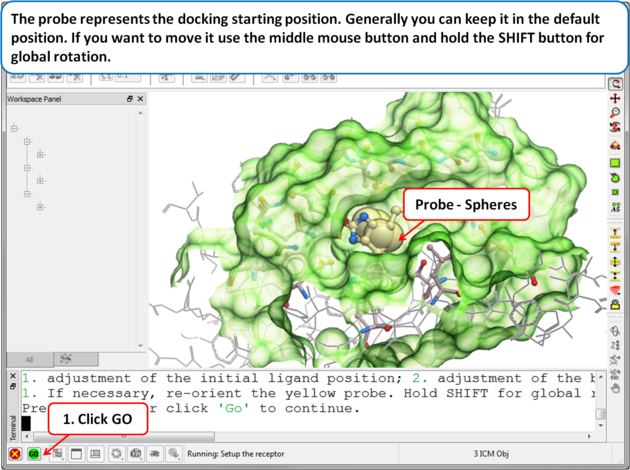 | |
| Step 6: Adjusting the Position of the Probe
The position of the probe (usually represented as 4 spheres in the center of the pocket) represents the initial position where sampling will begin. The default probe position is generally OK for most purposes but if you would like to move it to a critical part of the receptor so that sampling initially concentrates in that region you can do so using the middle mouse button and holding the SHIFT button for global rotation. Once you are happy with the position of the box press the enter key or click on "GO".
|
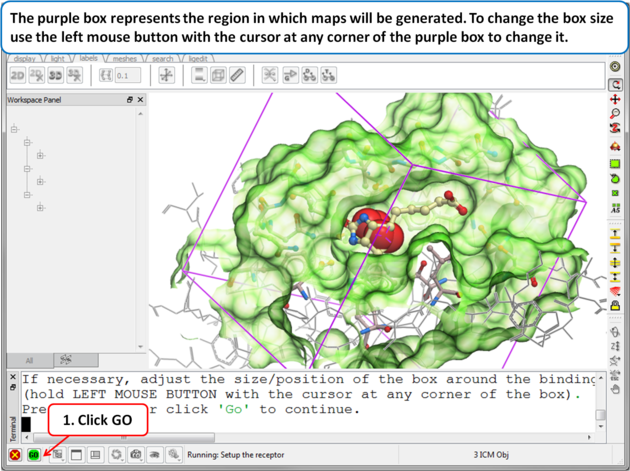 | |
| Step 7: Adjusting the Size of the Box
The purple box represents the region in which maps will be generated. The box needs to be large enough to encompass the binding pocket but not too large and including regions of the receptor which are not relevant for the ligand to bind. If the binding site is correctly defined in the earlier Receptor setup then the default box size is usually fine. If it is necessary to change the box size you can use the left mouse button with the cursor at any corner of the purple box to change it.
|
If at any point you want to change the binding site use the option Docking/Review Adjust Ligand/Box... Here you can change the initial position of the probe (default center or box) and the size of the box. If you change the size of the box you need to re-make the maps using the option Docking/(Re) Make Receptor Maps. You can use this option to also change the default map parameters such as the grid cell size and map weighting by occupancy.
If you change the size of the binding site box you will need to re-make the maps. To do this:
- Docking/(Re) Make Receptor Maps
[ General Preferences | Database Scan Preferences | Display Preferences ]
Usually the default preferences are sufficient, but several docking preference options are available in the Docking / Preferences menu. One important setting to consider is how ligands are charged. If you want to use MolSoft’s built-in pKa model, select the auto option under Docking / Preferences / General / Charge Groups.
Docking/Preferences/General
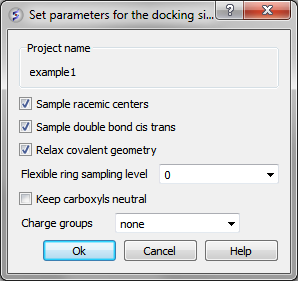
You can choose to:
- Sample racemic centers.
- Sample double bonds as cis trans.
- Relax covalent bond geometry (set as yes by default).
- Flexible ring sampling level. This option can be used for macrocycles. If the value is set to "1" then the ring is only flexible in pre-sampling. If the value is set to "2" then the ring is flexible throughout the simulation.
- Keep carboxyls neutral (set as no by default).
- Charge groups: provides simple interface to charge some ionizable basic groups, it currently understands following values: NH2, NH , NT for primary secondary and tertiary aliphatic amines, also it understands 'imidazole' and 'amidine'. If you have charged your ligands in a separate software you can use the option 'none' if not it is recommended to use the option 'auto', which uses built-in prediction of Ka/Kb to charge and protonate/deprotonate appropriate groups.
Docking/Prefences/Database Scan
These options are required when undertaking large scale docking and scoring using Virtual Ligand Screening (VLS). They are described in the VLS chapter of this manual.
Docking receptor-ligand display options can be changed in Docking/Preferences/Display
Now begin the docking procedure.