| Available in the following product(s): ICM-Pro | ICM-VLS | ICM-Chemist-Pro |
Video |
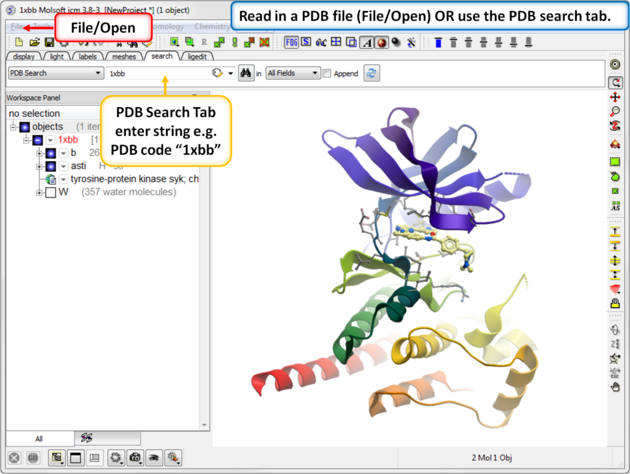
As an example we will use the kinase structure 1xbb.
- Click on the search tab and type 1xbb.
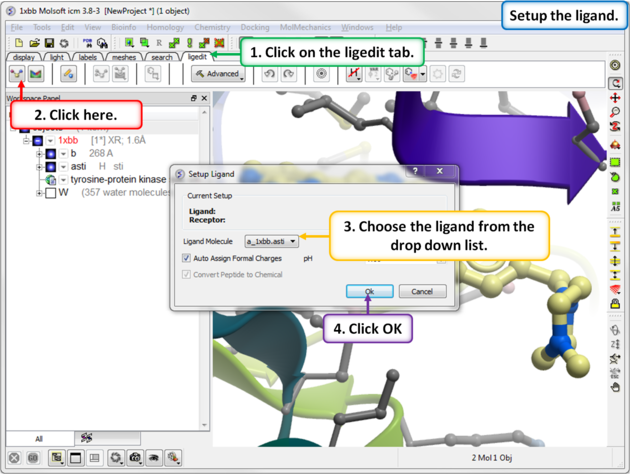
- Click on the ligedit tab
- Click on the Setup Ligand button.
- Enter the ICM selection language for the Ligand Molecule (a_1xbb_asti) or use the drop down button to locate it.
- Select whether or not you would like ICM to automatically assign formal charges and set the pH.
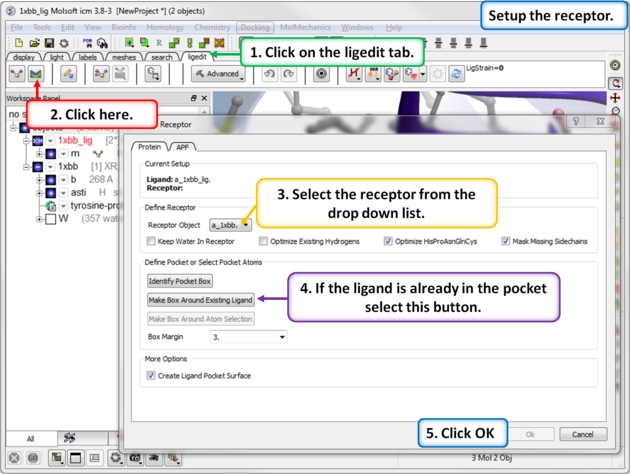
- Click the Receptor Setup button.
- Enter the ICM selection language for the Receptor Object (a_1xbb.), or use the drop-down menu to locate it.
- Choose your water and optimization preferences: keep all waters, keep tight waters, or delete all waters. You can also opt to optimize existing hydrogens and specific residues (His, Pro, Asn, Gln, and Cys). The "mask" option hides atoms not reported in the crystal structure (e.g., 0 occupancy); if left unchecked, ICM will automatically build the missing side-chain atoms. Read more about converting to an ICM object here.
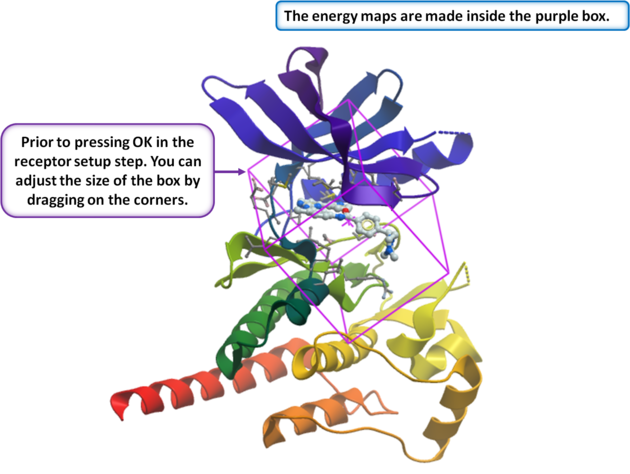
- Select Box Around Existing Ligand. Alternatively, choose Identify Pocket Box to run ICMpocketFinder, which returns a table of pockets—click your desired pocket in the table and press OK. A third option is to Make Box Around Atom Selection.
- Enter a box margin of 3. This defines the size of the energy maps around the ligand. A value of 3 typically encompasses the entire site; however, if your binding pocket is very elongated or unusual, visually verify that the purple box fully covers your region of interest.
- At this stage, you can resize the box by dragging its corners in the graphics display. The default size is generally appropriate, so changes are rarely needed. However, you should adjust it if the box does not completely cover the entire pocket, or if you plan to edit the ligand in a way that expands it into regions outside the current box.
- Click OK to generate the energy maps.
- Once everything is set up, you will see the ligand and receptor objects, along with the original PDB file, in the ICM workspace. At this point, you can save the complete setup as a project file (File/Save as .icb) so it is instantly available the next time you open it.
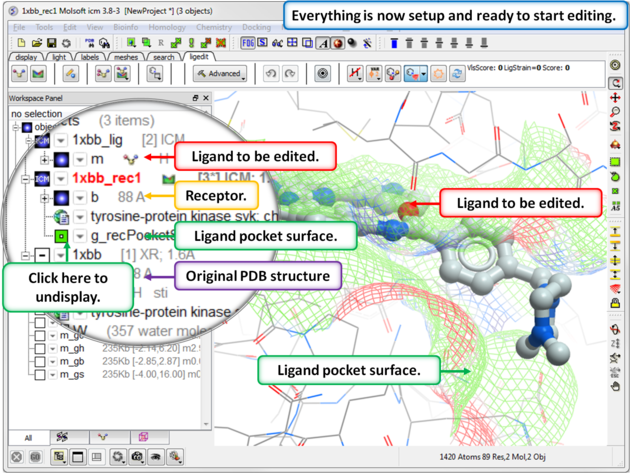
- Now everything is setup you can explore the pocket using the ligand editor display options and it is usually good practice to perform a short`re-dock-ligand{minimization} after setup.