| Prev | ICM User's Guide 9.20 Molecular Mechanics | Next |
[ ICM Convert | Optimize | Regularization | Impose Conformation | Edit Structure | MMFF | Torsion Scan | Minimize | Sample Loop | Design Loop | Sample Protein | Sample Peptide | Internal Coordinates Table | Normal Mode | Stack | GAMESS | Waters | Stack | Energy Terms ]
| Available in the following product(s): ICM-Pro |
9.20.1 ICM Convert |
To calculate energy, build a molecular surface and for all energy operations you need to convert a PDB file into an ICM object.
- MolMechanics/ICM-Convert/Protein
- Select the object you want to convert from the drop down list.
- Check or uncheck the options, delete water, optimize hydrogens, replace the original, and/or display the result.
To convert a small molecule into an ICM object.
- MolMechanics/ICM-Convert/Chemical
- Select the ligand.
- Choose whether you want to keep the current geometry of the ligand or not.
- Check or uncheck the options build hydrogens, fix amide bonds, and/or overwrite geometry.
9.20.2 Optimize H,His,Asn,Gln,Pro |
This option optimizes H, His, Asn, Gln, and Pro by maximizing hydrogen bonds and other interactions with the rest of the protein and/or with the ligand.
To perform this optimization
- Convert your protein to an ICM Object.
- Select MolMechanics/Optimize, H, His,Asn,Gln and Pro
- Choose whether you want to sample rotatable hydrogens or optimize His, Asn and Gln.
9.20.3 Regularization |
This option is described in detail in the modeling chapter here.
9.20.4 Impose Conformation |
If you have two protein structures with the same atom names and ALTER records but with different conformations you can impose the conformation of one of the protein structures onto the other.
You can do this by:
- Convert the structures into ICM Objects.
- MolMechanics/Impose Conformation
- Select the source molecule (the conformation you wish to impose).
- Optional: superimpose the structures
- Optional: re-optimize hydrogens
9.20.5 Edit Structure |
Set Bond Type
- Select the two atoms forming the bond.
- MolMechanics/Edit Structure/Set Bond Type
- Choose the Bond Type from the drop down arrow.
- Press Apply button
Set Formal Charge
- Select the atom.
- MolMechanics/Edit Structure/Set Formal Chage
- Choose the Charge from the drop down arrow.
- Press Apply button
Set Chirality
- Select the atom.
- MolMechanics/Edit Structure/Set Chirality
- Choose the Chirality from the drop down arrow.
- Press Apply button
Build Hydrogens
- Select the atoms.
- MolMechanics/Edit Structure/Build Hydrogens
- Press Apply button
Set Tether
Theory
A tether is a harmonic restraint pulling an atom in the current object to a static point in space. This point is represented by an atom in another object. Typically, it is used to relate the geometry of an ICM molecular object with that of, say, an X-ray structure whose geometry is considered as a target. Tethers can be imposed between atoms of an ICM-object and atoms belonging to another object, which is static and may be a non-ICM-object. You cannot create tethers in ICM-Browser, however, if the project that you have loaded contains tethers between two objects, then they can be displayed:
- Convert the two structures you wish to tether to an ICM object.
- MolMechanices/Edit Structure/Set Tether
- Right click on the first atom you wish to tether and click on the first option which is the selection language for the atom. This information will automatically be placed in the first atom dialog box. Alternatively you can type in the ICM selection language into the dialog box.
- Right click on the second atom (in a different object) you wish to tether and click on the first option which is the selection language for the atom. This information will automatically be placed in the second atom dialog box. Alternatively you can type in the ICM selection language into the dialog box.
- Press Apply button
Delete Tether
- Select the atoms you wish to delete the tethers from.
- MolMechanices/Edit Structure/Delete Tether
9.20.6 MMFF |
Set Types This option is described in detail here in the command line manual http://www.molsoft.com/man/icm-commands.html#set-type-mmff
Set Charges This option is described in detail here in the command line manual http://www.molsoft.com/man/icm-commands.html#set-chargemmff
Read Libraries This option is described in detail here in the command line manual http://www.molsoft.com/man/icm-commands.html#read-librarymmff
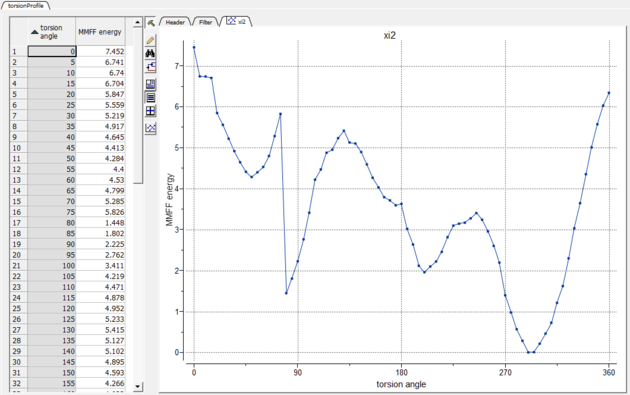
9.20.7 Torsion Scan |
The torsion scan is based on MMFF94s force field - A few corrections have been added over the years, most notably those from Wahl, Freyss, von Korff & Sander J of Cheminformatics 2019.
To sample the torsion angles.
- Read in or convert your molecule to an ICM object.
- Select the torsion angle. A convenient way of doing this is with the select atom selection tool.
- MolMechanics/Torsion Scan
- Enter the step in degrees you would like to sample.
- A table and plot will be displayed with the MMFF energy and angle.
9.20.8 Minimize |
Cartesian This option is described in detail here in the command line manual http://www.molsoft.com/man/icm-commands.html#minimize-cartesian
Local This option is described in detail here in the command line manual http://www.molsoft.com/man/icm-commands.html#minimize
Global This option is described in detail here in the command line manual http://www.molsoft.com/man/icm-commands.html#montecarlo
9.20.9 Sample Loop |
This option is described in the Loop Modeling section.
9.20.10 Design Loop |
This option is described in the Design Loop section.
9.20.11 Sample Protein |
This option is described earlier in this chapter here.
9.20.12 Sample Peptide |
This option is described earlier in this chapter here.
9.20.13 Internal Coordinates Table |
An easy way to set values of internal coordinates is to edit the Internal Coordinates Table directly in the GUI. Variables are discussed in more detail here: http://www.molsoft.com/man/selection.html#vs_
 |
| Step 1: The object you wish to edit has to be the Current Object. |
 |
| Step 2: Select MolMechanics/Internal Coordinates Table. Choose whether you wish to edit just the Free or All and include virtual variables. |
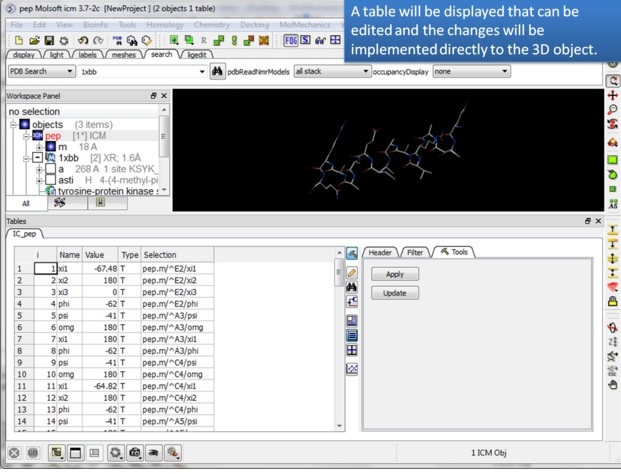 |
| Step 3: A table will be displayed containing the variable Name, Value, Type and Selection syntax. |
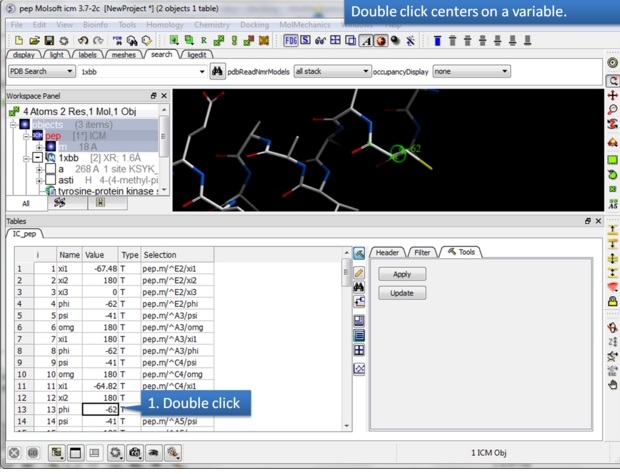 |
| Step 4: To center on a variable - double click on a row in the table. |
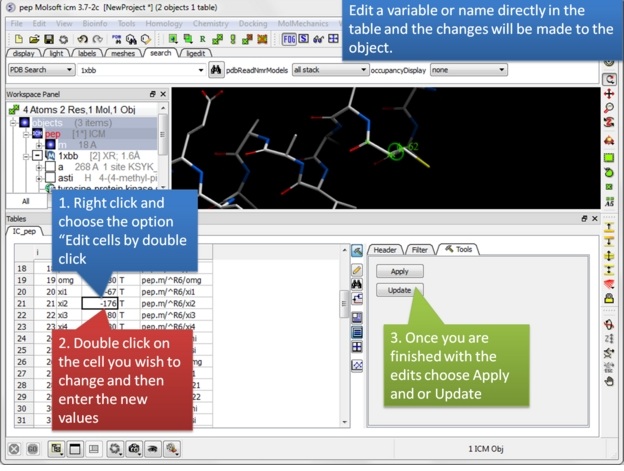 |
| Step 5: Edit a variable directly in the table and apply the change. |
9.20.14 Generate Normal Mode Stack |
Normal modes can be used to generate an ensemble of protein structures. For example the method can be used to represent flexibility in the pocket.
To generate an ensemble of structures using normal modes.
- Convert your protein to an ICM object.
- MolMechanics/Generate NM stack
- Enter the number of normal modes to sample
- Enter the relative amplitude of the normal modes.
- Optional: select to make random combination of modes.
- Optional: select Fast GAP model only.
- Optional: run the normal mode generation locally.
About Normal Modes:
Using multiple steps along the normal mode one can generate conformations with multiple amplitude values along each mode, so that larger and smaller movements for each mode are included in the stack. If no "interesting" movements are found in top 10 (slowest) modes, one can try to look at 20. If "interesting" movement happens to be 2 or 3, may be one need only look at top 3 or 5. There is absolutely no reason for the movement of interest to be at a particular rank as the protein may contain other mobile pieces in arbitrary places and numbers. The "interesting" movement may, unfortunately, also just not be there at all because not all movements are described well by the normal mode formalism. If one wants to analyze 'elementary' movements individually, it is best to look at pure modes; on the other hand in reality the molecule is moving along multiple modes simultaneously, therefore if a representative set of perturbed conformations is desired, randomly mixed modes may be more representative of the space of conformational perturbations accessible to a molecule.
Some papers describing Normal Modes in ICM:
9.20.15 Stack |
Operations which use the ICM Biased Probability Monte Carlo method e.g. docking and loop modeling generate a stack of energy conformations.
MolMechanics/Stack/View will display the conformations of a stack in a table ranked by energy. Each conformation can be viewed by double clicking on the table. A stack file will have the extension .cnf. For example, after running the sample loop algorithm a stack of different loop conformations will be generated.
MolMechanics/Stack/Play This option will play the elements of the stack as a movie. You can set the number of frames for the movie and also select whether you would like ICM to interpolate between each frame. You can save this movie in avi, mpeg format using the Screen-Grabbing Movie options.
MolMechanics/Stack/Add current conformation This option will add the currently displayed conformation to the stack. This is useful for experiments such as multiple receptor docking whereby you dock to a stack of conformations.
MolMechanics/Stack/Store Stack in Object This option takes the current stack and stores it in a compressed form inside the specified object. The compressed stack can then be extracted with the load stack object command. Option stack of the montecarlo command stores the generated stack inside the current object automatically.
To view the stack in gui:

MolMechanics/Stack/Delete Deletes the current stack.
MolMechanics/Stack/Set conf Comparison This option compares the stack as described here: http://www.molsoft.com/man/preference.html#compareMethod and http://www.molsoft.com/man/icm-commands.html#compare
MolMechanics/Stack/Recalculate Energies Recaluclates the energy of a current stack if changes have been made.
9.20.16 GAMESS |
This option is described in detail here in the command line manual http://www.molsoft.com/man/E-H.html#gamess
You need to register on the GAMESS website and submit download request:
https://www.msg.chem.iastate.edu/GAMESS/
Few notes:
- Make sure to specify full path gamess in the dialog. (default: c:/Users/Public/gamess-64/rungms )
- Make sure to edit rungms script and set default version. Find 'SET VERSION' line and set the default according to you setup e.g. 2019.R1.P1.mlk
- It is a good idea to set NCPUS to something higher than 1 to speed up calculations. Search for 'SET NCPUS'
9.20.17 Waters |
'Flood' procedure populates a box around the region of interest on the protein with water molecules, generating a stack of low-energy water configurations. 'Quick flood' performs the procedure within the interactive ICM session. For more thorough sampling, it is recommended to use 'Sample Flood' that runs the same procedure in the background.
Try an example:
- MolMechanics/Waters/Load Example
- Click on the html link to generate a box around the region of interest. You should see a purple box - the size of it can be changed by dragging on the corners of the box. In a real example you can make a selection and then in the MolMechanics/Waters/Sample Flood dialog box there is a button "Make Box Around Selection".
- MolMechanics/Sample Flood
- Enter the name of the protein.
- Enter the number of water configurations you would like to generate.
- Choose whether to make the water density map and/or contour.
- Click OK.
- A stack of low energy water molecule configurations will then be displayed in the ICM Workspace.
9.20.18 Working with Stacks |
Many modeling functions in ICM return a stack of conformations. For example:
- Loop Modeling
- Stack for Multiple Receptor Conformation(4D)docking and screening
- Molecular Dynamics returns a stack of structure snapshots over time.
ICM has a variety of tools to analyze and make calculations based on the stack. Right click on the stack in the ICM workspace and choose Stack Calculations.
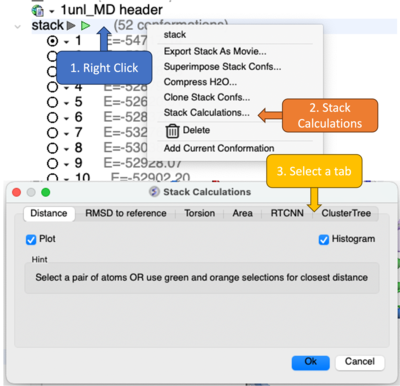
A dialog box will be displayed providing analysis tools as described below.
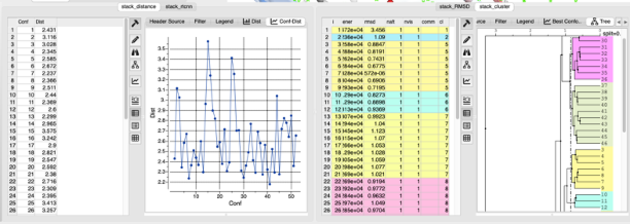
- Distance - Select a pair of atoms or use green and orange selections for closest distance. A table and plots of the distance will be displayed.
- RMSD to Reference - Read in a separate object as a reference and make the selection you want to analyze. You can choose to analyze the structures 'static' or superimpose. A table and plot of the RMSDs are displayed. This is a useful tool for analysis of a Molecular Dynamics run. Your x-axis will be the time in ns / number of conformations so if you chose 1ns and 50 conformations (snapshots) then each point is 1ns/50 = 0.02 ns
- RMSF - Root mean square fluctuation indicates positional differences in structure during a simulation. There is a Static and Superimpose option - the superimpose option follows protocol described here. You can provide two selections: for superposition using orange selection and regular "green" selection for RMSF calculation. By default it (if no selections are provided) backbone from all residues will be used.
- Ligand Contacts - This method uses our Interaction Lists macro from our docking hitlist. Select the ligand and then the macro will be called for each MD snapshot in the stack and calculates number of: hydrophobic(apol), hydrogen bond don/acc (hba/hbd) and positive/negative (pos/neg) - positive or negative (formal charge) atom in contact (3.5 - 4.5A) with receptor residue and one ligand atom can participate in more than one interaction.. Individual residues can be included if you check this option.
- Torsion - Select a pair of atoms on a rotatable bond to calculate the torsion angle.
- Area - use green and orange selections to calculate differences in area.
- RTCNN - use green and orange selections for receptor/ligand respectively to calculate RTCNN docking score.
- ClusterTree - make a selection of atoms to compare and then a cluster tree will be displayed. You can then 'select-tree{select} a diverse set of conformations in the tree.
The results from the calculations are presented in a table along with the associated plots and trees.
9.20.19 Energy Terms |
The energy function calculated for any conformation of an ICM molecular object consists of individual terms described which can be turned on and off using MolMechanics/Energy Terms. These terms are described in more detail here http://www.molsoft.com/man/terms.html
| Prev Sample Peptide | Home Up | Next Homology Modeling |